
Malattia del Motoneurone Malattia di Charcot Malattia di...
132
Sclerosi Laterale A miotrofica Malattia del Motoneurone Malattia di Charcot Malattia di Lou Gehrig Dott.ssa Romana Rizzi Dott.ss Elena Canali ASMN Neurologia
-
Upload
doankhuong -
Category
Documents
-
view
216 -
download
0
Transcript of Malattia del Motoneurone Malattia di Charcot Malattia di...

Sclerosi Laterale Amiotrofica
Malattia del MotoneuroneMalattia di Charcot
Malattia di Lou Gehrig
Dott.ssa Romana RizziDott.ss Elena CanaliASMN Neurologia

Malattie del Motoneurone
Sono una serie di malattie che hanno in comune la degenerazione e la morte
cellulare dei neuroni motori:
SLASclerosi Laterale PrimariaAtrofia Muscolare Spinale
Malattia di KennedySdr. Di Vialetto
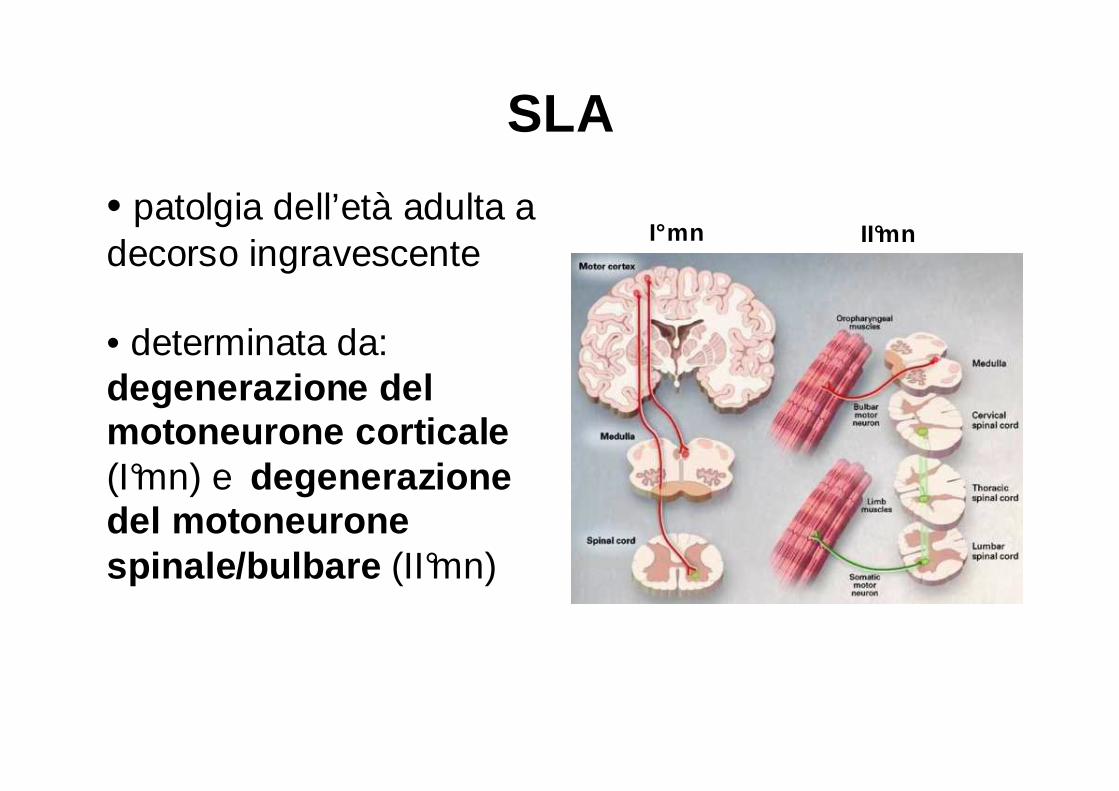
SLA
• patolgia dell’età adulta a decorso ingravescente
• determinata da: degenerazione del motoneurone corticale (I°mn) e degenerazione del motoneurone spinale/bulbare (II°mn)
I°mn II°mn
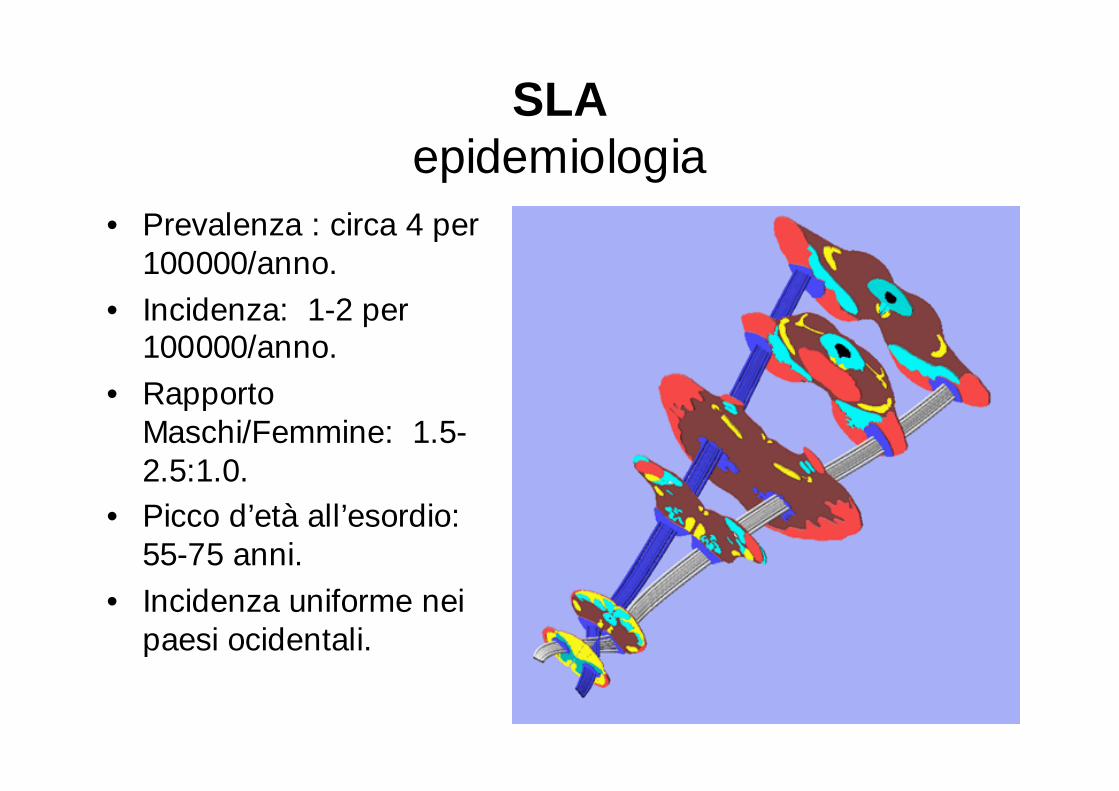
SLAepidemiologia
• Prevalenza : circa 4 per 100000/anno.
• Incidenza: 1-2 per 100000/anno.
• Rapporto Maschi/Femmine: 1.5-2.5:1.0.
• Picco d’età all’esordio: 55-75 anni.
• Incidenza uniforme nei paesi ocidentali.

SLA: eziopatogenesi
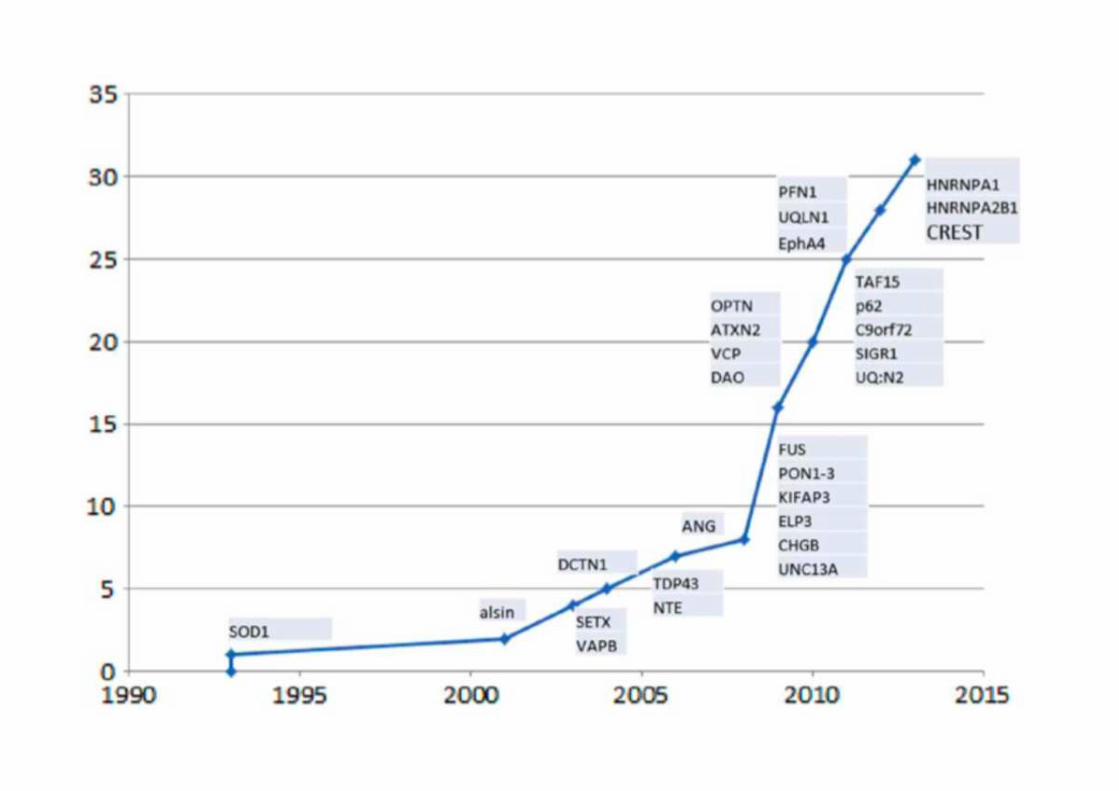
La scoperta della mutazione nel gene SOD1, sito sul cromosoma 21q22.11, ha dato il maggior contributo ad un’iniziale comprensione dei meccanismi patogenetici della SLA.
La perdita di funzione di SOD1 determina un effetto tossico neuronale, sia a livello cellulare che nucleare venendo a meno la protezione del DNA dall’effetto dei radicali liberi (ipotesi del danno ossidativo ).
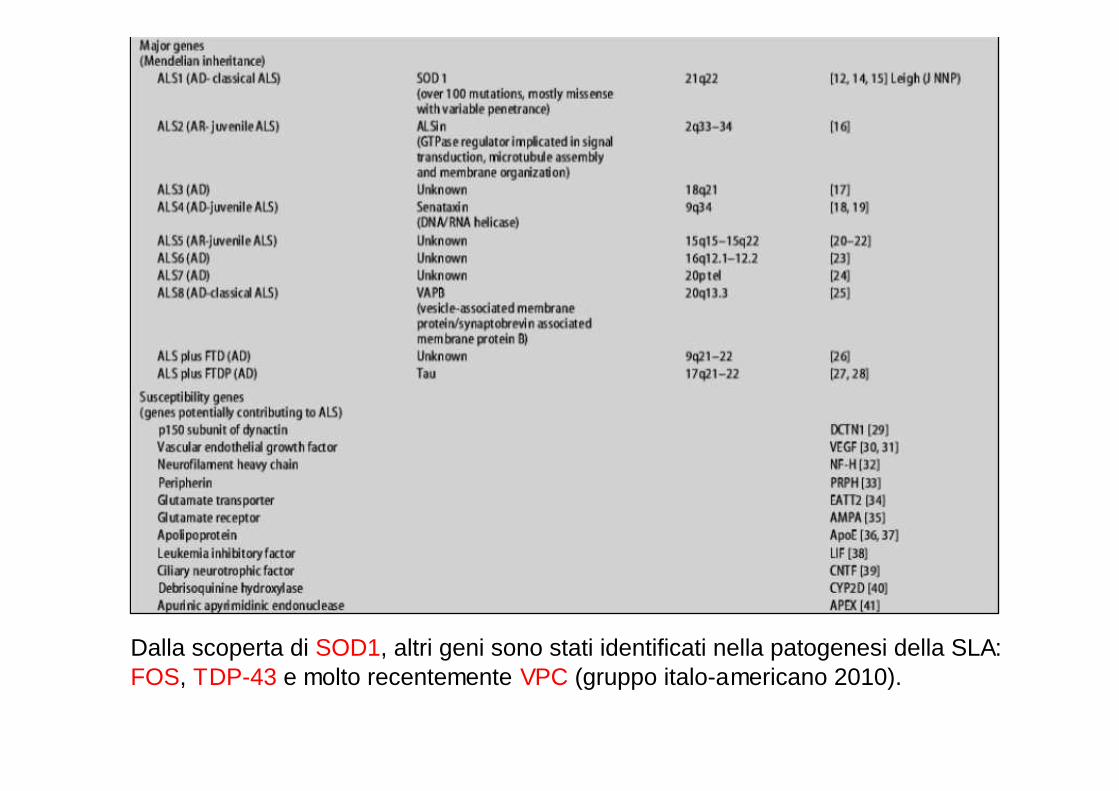
Dalla scoperta di SOD1, altri geni sono stati identificati nella patogenesi della SLA:FOS, TDP-43 e molto recentemente VPC (gruppo italo-americano 2010).
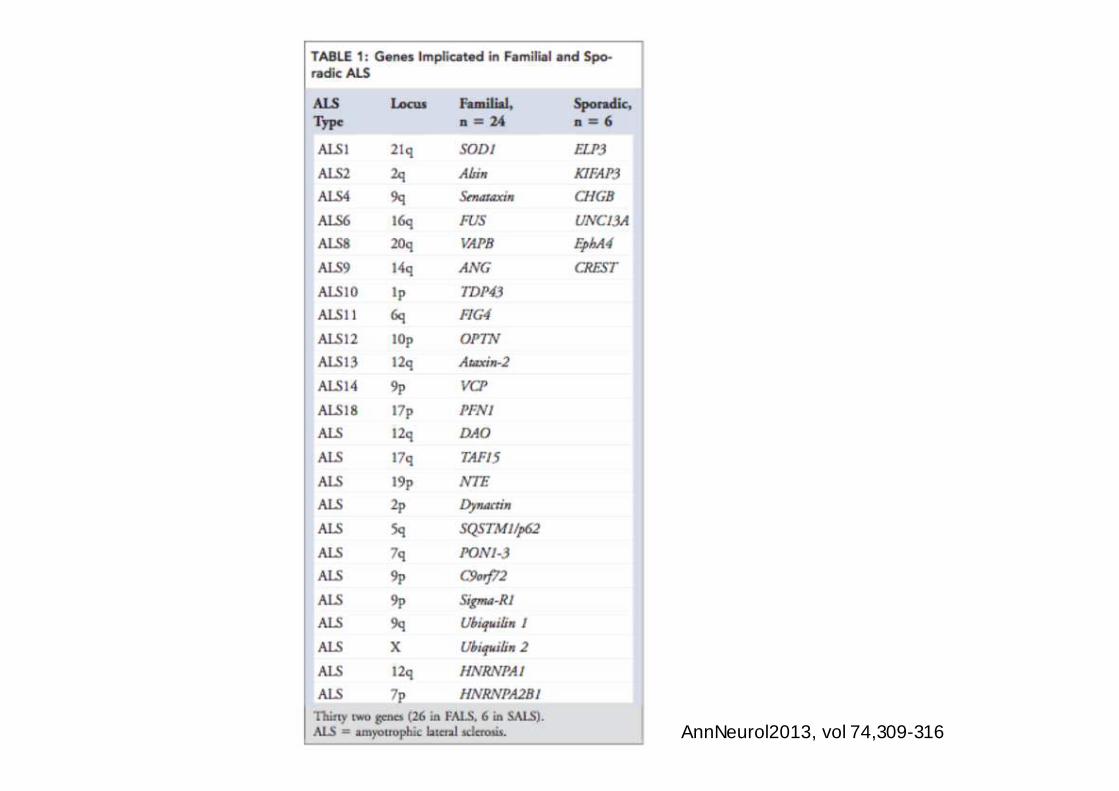
AnnNeurol2013, vol 74,309-316
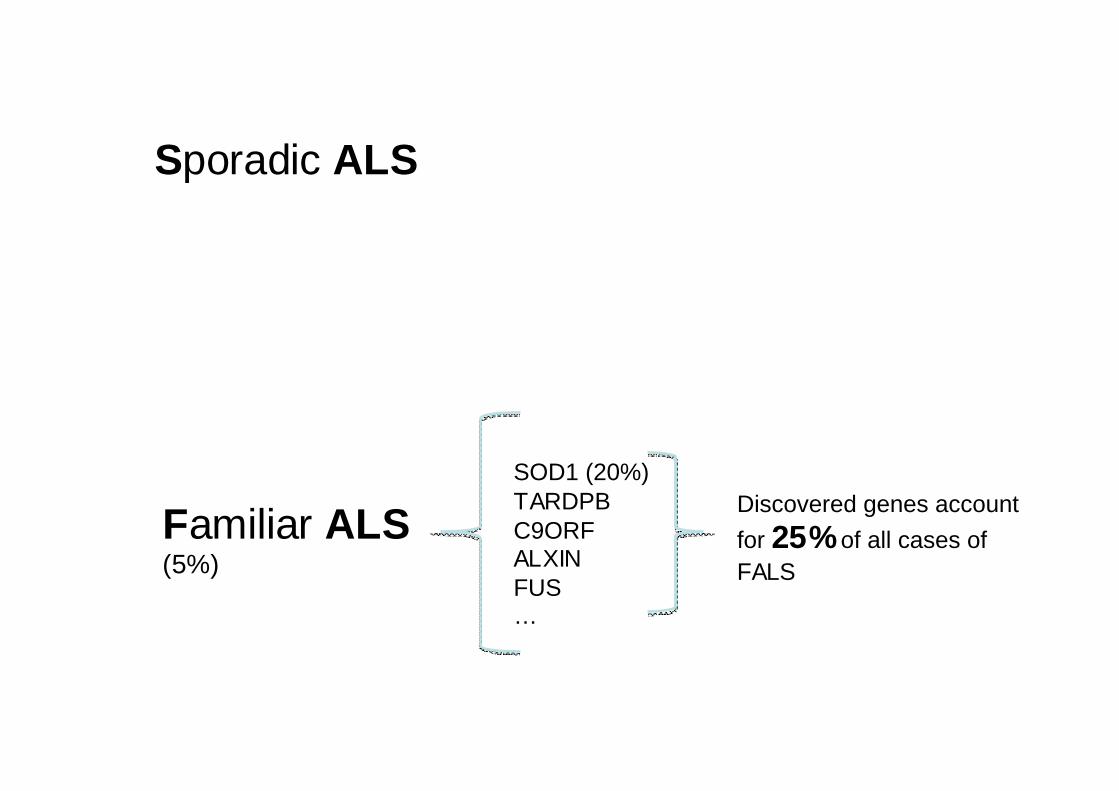
Sporadic ALS
Familiar ALS(5%)
SOD1 (20%)TARDPBC9ORFALXINFUS…
Discovered genes account
for 25%of all cases of FALS

Locus Gene N.mutations Phentype Other features
ALS1 SOD1 166 ALSPMA
Cognitive impairment rare
ALS2 ALS2 19 Juvenile ALS unknown
ALS3 Not identified unknown ALS unknown
ALS4 SETX 9 ALS Cerebellar ataxia
ALS5 SPG11 12 Juvenile ALS unknown
ALS6 FUS 42 ALS-FTD FTDPArkinsonism
ALS7 Not identified unknown ALS unknown
ALS8 VAPB 99 ALS,PMA unknown
ALS9 ANG 17 ALS,ALS-FTD
Parkinsonism
ALS10 TARDBP 44 ALS-FTD PSP,PD,FTD
ALS11 FIG4 10 ALS,PLS CMT4J, cognitive impairment
ALS12 OPTN 5
ALS13 ATXN2 6 ALS unknown

ALS and FrontoTemporal Dementia:the c9ORF era
• espansione esanucleotidica GGGGCC• responsabile di più del 30% di SLA familiari• scoperta del gene nel 2011• associazione ALS-Fronto Temporal Dementia
Fenotipo clinico FALS c9ORF• età di esordio variabile (27-83)• più rapida evoluzione di malattia• il 29,3% dei pazienti presenta sintomi di ALS + FTD• 44,2% esordio bulbare (>rispetto alla media)• associazione di parkinsonismo, psicosi

SLA: eziopatogenesi
Alla luce delle evidenze di cui si dispone,la SLA sembra rappresentare una patologia a genesi multifattoriale:
• substrato genetico predisponente?• fattori ambientali? (danno ossidativo da esposizione a tossici, stili di vita, lavoro..)• altro?

Patogenesi del dannoIl danno nella forma più classica
inizia con il coinvolgimento delle corna anteriori del midollo spinale : i neuroni vanni in apoptosi ovvero incontro alla degenerazione e alla morte.
Contemporaneamente o in un secondo momento, anche i motoneuroni corticali vanno incontro a patologia .
I motoneuroni dei nervi cranici vago, accessorio e ipoglosso sono frequente colpiti, anche se la distribuzione del danno è irregolare.
II°MOTONEURONE
iposteniaatrofia muscolarefascicolazioni muscolari
ipofoniadisfagiadisartria
I°MOTONEURONE
Iposteniaiperreflessia segno di Babinskispasticità (più rara)

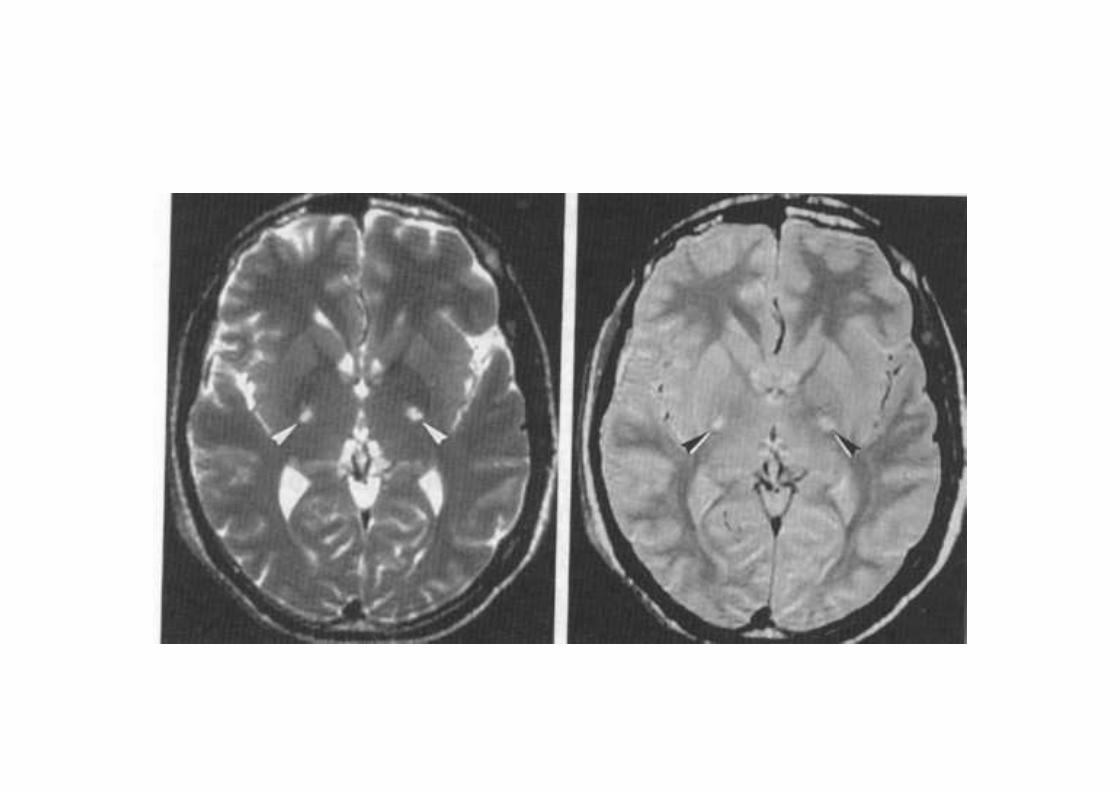
I fasci piaramidali di dx e di sx possono essere di fferentementeinteressati dalla perdita di fibre, le aree maggior mente colpitesono la zona dei peduncoli cerebrali e la zona dell a capsula interna. Le lesioni dei fasci piramidali sono visibili con l a RM apparendo come segnali iperlucenti in T2.

Quadro clinico
Degenerazione I°mn (sdr.piramidale): ROT vivaci, segno di Babinski (50%dei casi), riflesso masseterino.
Degenerazione del mn bulbare :disartria, disfagia, labilitàemotiva (sdr.pseudobulbare). In alcuni pz la compromissione bulbare è nettamente dominante e il distretto cranico rimane a lungo l’unico interessato.
Degenerazione II°mn : scomparsa dei ROT (nella 2°fase di malattia), fascicolazioni (a volte precoci), atrofia muscolare.

Quadro clinico
Assenza di sintomi sensitivi (dolore…).
Non deficit della muscolatura oculare estrinseca.
Non disfunzioni sfinteriali.
Generalmente non lesioni da decubito.

Quadro clinico
La degenerzione dei motoneuroni è progressiva e determina ipostenia e ipotrofia che si estende a tutta la muscolatura corporea.
L’exitus del paziente avviene in media entro 36 mesi dall’esordio dei sintomi, generalmente per insufficienza respiratoria , polmonite ab ingestis o malattie infettive ricorrenti .
Il trattamento invasivo dell’insufficienza respiratoria tramite ventilazione meccanica , previa tracheotomia, prolunga nettamente la sopravvivenza, che può superare in molti soggetti i 10 anni di vita.

la maggior parte dei casi sono SPORADICI
10% dei casi FAMILIARITA’ : un gene identificato nella forma tipica di SLA è quello
che codifica per l’enzima superossido dismutasi Cu/Zn (SOD-1), sito sul cromosoma 21, la cui mutazione è associata a circa il 15-
20% dei casi di SLA familiare.
SLAepidemiologia
Altri geni identificati: FOS, TDP-43, VPC…

Quadro clinico
Una variante clinica, più rara, è quella associata a decadimento cognitivo, con un quadro clinico tipo demenza frontotemporale (FTD) .
La frequenza di FTD associata a SLA varia dal 5 al 15% e sono state identificate alcune forme di SLA ereditarie caratterizzate da questa associazione.

Variabilità del quadro clinico
• Molti pazienti presentano soltanto segni di II motoneurone (amiotrofia) o solo di I motoneurone (spasticità) all’inizio della malattia.
• Negli arti la malattia può iniziare in maniera asimmetricasuccessivamente la malattia si estende a tutti i muscoli.
• La velocità di progressione della malattia è lineareanche se variabile da soggetto a soggetto (da forme letali in un anno a forme che evolvono lentamente).

Diagnosi
1. Anamnesi
2. Esame obiettivo
3. Elettromiografia (EMG)
Non esiste un marker biologico di malattia !

Diagnosi
Anamnesi, esame obiettivo ed EMG
mirano alla ricerca di segni neurologici di sofferenza sia del I°sia del II°motoneurone in 4 regioni :
Distretto cranico (mandibola, palato, lingua, laringe)
Distretto cervicale (nuca, AASS, mani, diaframma)
Distretto toracico (tratto dorsale e addome)
Distretto lombosacrale (tratto L-S, addome,AAII e piedi)

I criteri diagnostici di El Escorial
• SLA clinicamente definita: presenza di segni sia di I sia di II mnnella regione bulbare e in almeno due regioni midollari oppure in almeno tre regioni midollari (cervicale, toracico, lombosacrale).
• SLA probabile : presenza di segni di I e di II motoneurone in almeno due regioni (con alcuni segni di I rostrali a quelli di II)
• SLA possibile: presenza di segni di I e di II motoneurone insieme in solo una regione o segni di I da soli in due o più regioni, o segni di II rostrali a segni di I.
• SLA sospetta: è una sdr del II mn pura. Questa categoria è stata esclusa dalla revisione dei criteri di El Escorial.

1990 “El Escorial Diagnostic Criteria”
2000 “Revised El Escorial Diagnostic Criteria” (Airlie House Criteria)
2006 Awaji – Shima Consensus Conference
Diagnosi Neurofisiologica: cosa è cambiato?

Clinical diagnostic certainty :The Airlie House Criteria (2000)
Revised El-Escorial (2000)Revised El-Escorial (2000)
Clinicamente possibile
Clinicamente probabile
con supporto di laboratorio
Clinicamente probabile
Clinicamente definita
UMN + LMN in 1 regione
oUMN in 2 regioni
oLMN rostrali a
UMN
UMN + LMN in 1 regione / UMN in >1 regione
+EMG denervation
> 2 limbs
UMN +
LMN in 2 regioni
UMN +
LMN In 3 regioni

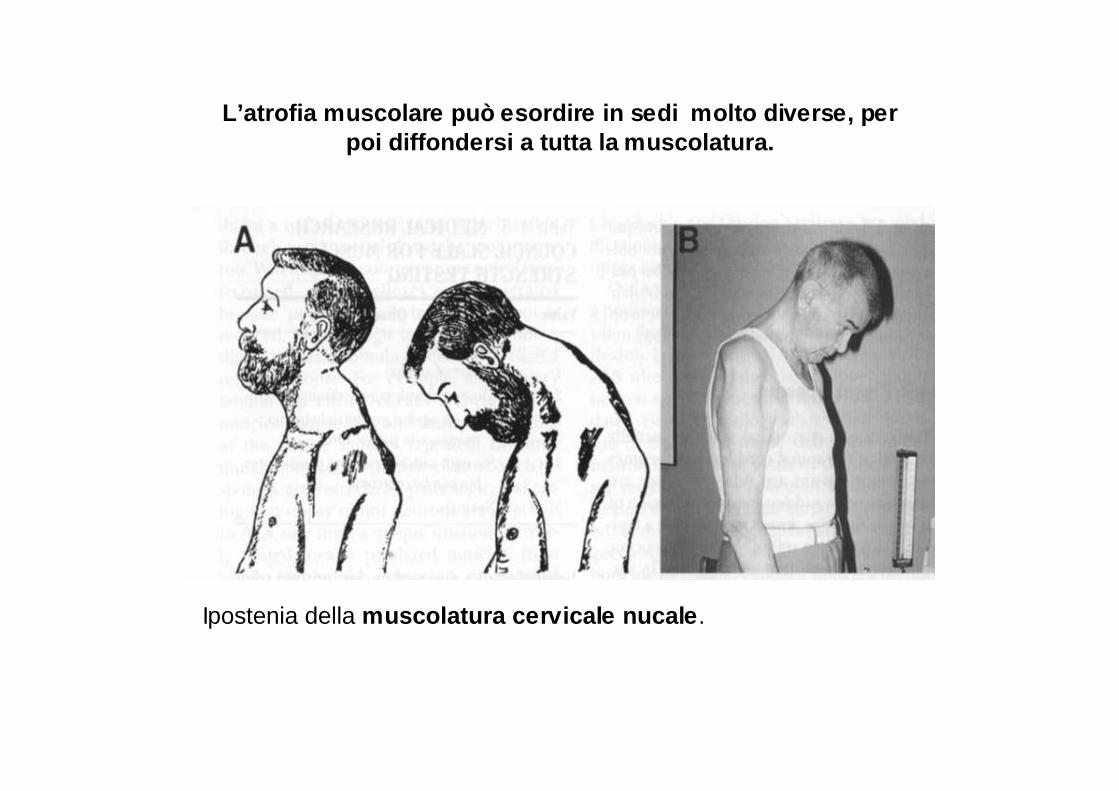
L’atrofia muscolare può esordire in sedi molto div erse, per poi diffondersi a tutta la muscolatura.
Ipostenia della muscolatura cervicale nucale .

Ipotrofia dei piccoli muscoli delle mani.
atrofia del m. I°interosseo dorsale.

SLA: diagnosi differenziale
• esclusione di altri segni elettrofisiologici o processi patologici che possano spiegare la degenerazione del I e del II MN (El Escorial criteria)
• neuropatia motoria multifocale, ipertiroidismo, ipercalcemia.
• mielopatia spondilosica, radicolopatie, post-polio sdr.

Diagnosi differenziale:SMA
Le atrofie muscolari spinali (SMA) includono un gruppo di patologie ereditarie caratterizzate da degenerazione delle cellule delle corna anteriori e clinicamente da ipostenia e atrofia muscolare.L’età di esordio è variabile, in genere nell’infanzia per la SMA tipo I e tipo II e nell’adolescenza per la SMA tipo III e tipo IV.
L’ipostenia e l’atrofia sono simmetriche e interessano prevalentemente la musolatura prossimale. La SMA I e II hanno un decorso rapidamente progressivo che porta a morte nei primi anni di vita. La SMA III e IV hanno un’evoluzione molto più lenta e prognosi favorevole qoad vitam.

Diagnosi differenziale:neuronopatia bulbospinale
(M.di Kennedy)
• Malattia ereditaria legata al cromosoma X, causata dall’espansione della tripletta CAG nel gene per il recettore degli androgeni.
• esordio nell’età adulta, ginecomastia, tremore posturale, faticabilità.
• successiva comparsa di atrofia simmetrica prossimale, segni bulbari (disfagia), ipostenia facciale.
• non segni di I°mn.
• alterazioni ormonali, CPK elevato.
• alterazione dei SAP all’EMG.
• progressione molto lenta.

Principi generali di trattamento
Il cardine del trattamento nei pazienti affetti da SLA rimane iltrattamento sintomatico.
• importanza di affiancare al malato un team multidisciplinare di specialisti (neurologo, pneumologo, dietologo, psicologo, fisioterapista,terapista occupazionale, foniatra, infermiere etc…)
• impostare un corretto rapporto medico-paziente-famiglia (il paziente deve essere informato sulla diagnosi e sull’evoluzione per scegliere in modo consapevole circa la propria assistenza)

Principi generali di trattamento
Patients with ALS receiving multidisciplinary team care live significantly longer compared
to patients in neurology clinics.
Bedlack CurrOpin Neurol 2010

Principi generali di trattamento
Terapia farmacologica specifica
Il Riluzolo è l’unico farmaco attualmente approvato in Europa e USA per il trattamento della SLA.Effetto antiglutammatergico, riduzione dell’eccitotossicità.Uno studio condotto in doppio cieco verso placebo su 959 pz ha dimostrato un’aumento della sopravvivenza di circa 3 mesi nei pz trattati.
Posologia: 50mg 2v/die.
Effetti coll: nausea, vertigini, disturbi gastrointestinali (5% dei pz), rialzo transaminasi.
Una review recente non ha dimostrato l’efficacia della vitamina E come supporto alla terapia farmacologica. (Miller et al. Neurology 2009)

Principi generali di trattamento
Terapia farmacologica sintomatica
• crampi e fascicolazioni : chinino solfato, magnesio, baclofen, fenitoina.
• spasticità: baclofen, diazepam.
• scialorrea : amitriptilina, scopolamina.
• depressione : amitriptilina, venlafaxina, mirtazapina…
• ansia : lorazepam, diazepam, alprazolam..
• minzione imperiosa : ossibutinina, amitriptilina.
• insonnia : zolpidem..
• stipsi : lattulosio, senna..

I BISOGNI DEL PAZIENTE
MOTRICITA’MOTRICITA’
COMUNICAZIONECOMUNICAZIONE
RESPIRAZIONERESPIRAZIONE
NUTRIZIONENUTRIZIONE
FISIOTERAPIA
AUSILI
LOGOTERAPIA
COMUNICATORI
NUTRIZIONISTA
PEGSUPPLEMENTI DIETETEICI
PNEUMOLOGO
NIV
TRACHEOTOMIA
FISIOTERAPIA RESPIRATORIA
TERAPIA OCCUPAZIONALE
RIG (radiologically inserted gastrstomy)


Disartria : il logopedista può aiutare il paziente insegnando tecniche per articolare la parola, fino a quando il peggioramento del quadro clinico porta il paziente all’anartria e diventa necessario fornire e insegnare l’uso del PC o di un comunicatore al paziente.
COMUNICAZIONECOMUNICAZIONE

Disfagia : le difficoltà della deglutizione intevengono al momento del coinvolgimento dei neuroni bulbari dei nervi cranici V (Trigemino motore), X (vago)-XI(accessorio) e XII (ipoglosso): inizialmente il paziente rallenta la masticazione e presenta episodi di disfagia (anche con la sola saliva) ma aggravandosi i sintomi di ab ingestiis la nutrizione per os diviene molto pericolosa per cui è necessario l’impianto della PEG (Percutaneous Endoscopic Gastrostomy) che può migliorare la qualità e prolungare la vita di qualche mese.
NUTRIZIONENUTRIZIONE
la PEG e la NUTRIZIONE
1. La PEG aumenta la sopravvivenza dei pazienti con SLA. Il problema è decidere quando posizionarla: il rischio legato alla procedura aumenta quando l’FVC scende sotto al 50%.
2. Dieta lipidica : alcuni studi recenti hanno dimostrato l’efficacia dei lipidi sulla sopravvivenza dei pz con più alti livelli di Colesterolo LDL.

la PEG e la NUTRIZIONE
Un’alternativa alla PEG, considerata una manovra più sicura nel paziente con iniziale deficit respiratorio, è la RIG: Radiologically Inserted Gastrostomy (il device viene inserito dal radiologo “rx guidato”, senza necessità di gastroscopia).
La nutrizione enterale (PEG) è efficace nello stabilizzare il BMI (2 studi di classe II) (Neurology 23, 2009)
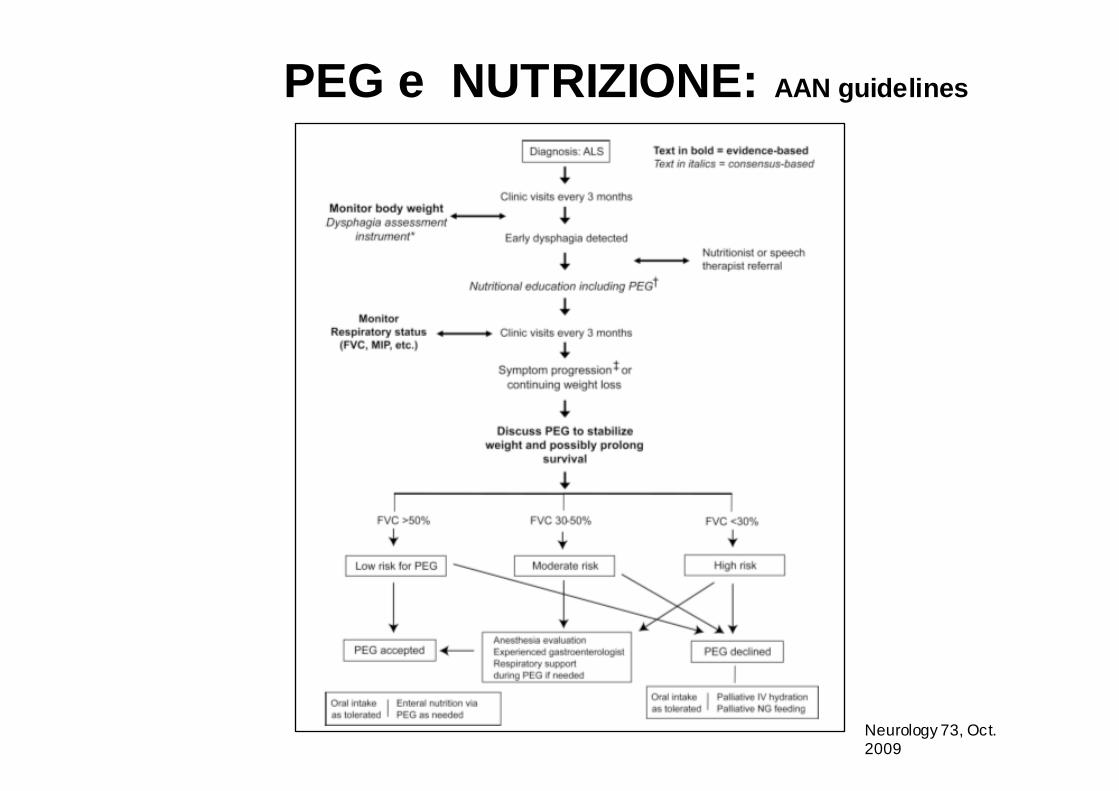
PEG e NUTRIZIONE: AAN guidelines
Neurology 73, Oct. 2009

Gli studi che hanno usato controlli appropriati ed analisi multivariate hanno dimostrato che la PEG è efficace nel prolungare la sopravvivenza dei pazienti affetti da SLA (2 studi di classe II).
Neurology 73, October 13, 2009

Salivazione: normalmente produciamo 500 ml di saliva nelle 24 ore. Con l’aggravarsi della difficoltà di deglutizione il paziente presenta scolo di saliva per cui si rende necessario fornire alcuni farmaci anticolinergici come l’amitriptilina (Laroxyl) da 10 a 100 mg. Altri farmaci possono essere la scopolamina anche in cerotto.
Spasticità: a causa della sindrome piramidale il paziente presenta crampi e rigidità : sintomi spesso dolorosi. Il baclofen e il diazepam sono i farmaci di prima scelta.
Costipazione : può essere una conseguenza della debolezza dei muscoli pelvici e addominali, per la diminuita attività fisica, per le medicazioni colinergiche e antispastiche e gli oppioidi.

Ginnastica respiratoria: è importante per mantenere l’elasticità polmonare, per ridurre il ristagno di secrezioni bronchiali e quindi il rischio di infezioni respiratorie.
Terapia fisica : l’attività fisica è importante per il mantenimento dell’articolarità mediante esercizi fisici di stretching, passivi e attivi per prevenire le contratture.
RESPIRAZIONERESPIRAZIONE
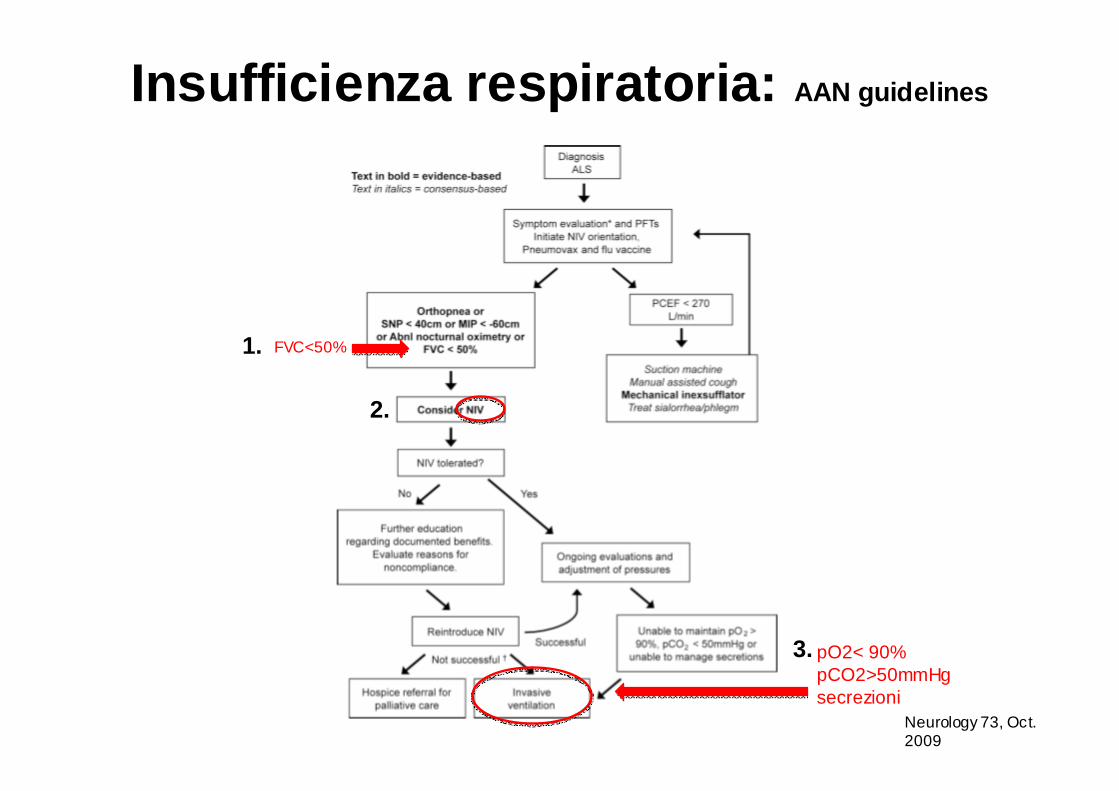
Insufficienza respiratoria: AAN guidelines
Neurology 73, Oct. 2009
FVC<50%
pO2< 90%pCO2>50mmHgsecrezioni
1.
2.
3.

• l’ossimetria notturna è raccomandata per valutare l’ipoventilazione (AAN 2009) e iniziare la NIV (indicata se Pa02<90% per 5% del sonno).
• l’FVC in paziente supino è più efficace nel valutare la funzione diaframmatica e correla meglio con i sintomi di ipoventilazione notturna.
• la NIV è probabilmente efficace nel prolungare la sopravvivenza e nel rallentare il declino dell’FVC (Neurology 73; Oct 13,2009).
• la NIV migliora la qualità della vita dei pazienti con SLA (maggior energia diurna, vitalità, minor sonnolenza, miglior qualità del sonno, meno depressione…)
• l’uso di Mechanical Insufflation/exsufflation (cough machine) è utile per rimuovere le secrezioni.

Sintomi indicativi di ipoventilazione notturna
Insonnia (di addormentamento, di mantenimento, risvegli)
Arousal prolungati
Sensazione di sonno non riposatoreIncubi
Scarsa energia diurnaSonnolenza diurna/scarsa concentrazioneDepressione
Cefalea mattutinaScarso appetito

Criteri per intraprendere NIV (Non Invasive Ventilation)

Tipologie di ventilatori (Non Invasive Ventilation)
nasal
facetotal face
helmet
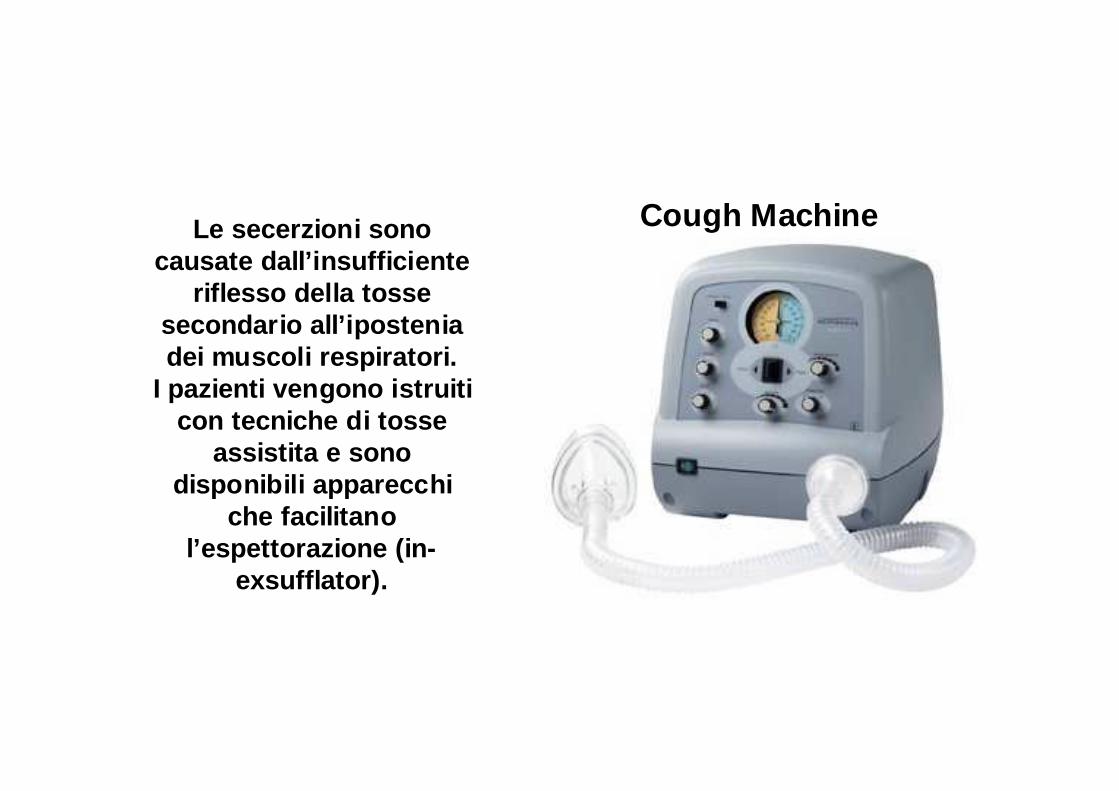
Cough MachineLe secerzioni sono causate dall’insufficiente
riflesso della tosse secondario all’ipostenia dei muscoli respiratori.
I pazienti vengono istruiti con tecniche di tosse
assistita e sono disponibili apparecchi
che facilitano l’espettorazione (in-
exsufflator).

NIV: compliance
• abituarsi all’uso del ventilatore per il paziente è difficile e non tutti riescono.
• la compliance del pz alla NIV migliora se viene iniziata precocemente.
• i pazienti con SLA bulbare o con SLA + demenza FT presentano minor compliance.
• una volta abituati, i pazienti sperimentano una miglior qualità del sonno e una maggior forza muscolare dovuta alla miglior ossigenazione tissutale.

L’ossigenoterapia pura , finalizzata a correggere l’ipossiemia, va utilizzata con molta cautela per i l rischio di peggiorare drasticamente la PaC02 e causare coma ipercapnico .Trova il suo impiego soprattutto nel trattamento dell’ipossia nelle fasi terminali.

SLA: scelte di fine vitaQuando la NIV non è più efficace, è necessario decidere se
proseguire con la ventilazione invasiva (tracheotomia) o solo con un approccio palliativo nelle fasi terminali.
La scelta della ventilazione invasiva deve essere preceduta da un approfondito dialogo medico-paziente-famiglia per le pesanti ripercussioni psicologiche, assistenziali e economiche per il paziente e la sua famiglia.
Gli studi sulla ventilazione invasiva mostrano una sopravvivenza a 5 anni del 25-35%.
La morte di solito avviene per infezioni respiratorie o èimprovvisa.

SLA: scelte di fine vita
In Italia al paziente in NIV è consentito esprimere le “direttive anticipate” in cui esprimere proprie volontà in caso di improvvisa insorgenza di insufficienza ventilatoria:
1) Soccorso rianimatorio con intubazione e successiva tracheotomia.
2) Rifiuto della tracheotomia. Terapia palliativa (oppioidi) per alleviare i sintomi dell’insufficienza ventilatoria fino all’exitus.
Tali decisioni sono modificabili in qualsiasi momento.
E’ auspicabile che il paziente le esprima per iscritto.

SLA: scelte di fine vita
In molti paesi europei (NON in Italia) e negli USA la legislazione vigente permette al paziente di esprimere attraverso direttive anticipate la volontàche vengano interrotte le misure di supporto vitale in qualunque momento nel corso della ventilazione meccanica.

considerazioni…
È importante una corretta informazione e formazione di tutti gli operatori sanitari sulla gestione del paziente con SLA per diffondere l’informazione che si può fare molto per alleviare le sofferenze dei pazienti.

considerazioni…
È importante chela diagnosi di malattia sia il più precoce possibile per permettere ai pazienti di essere arruolati nei trial terapeutici.
L’alleanza medico-paziente-infermiere èdecisiva nelle scelte del paziente e nell’influenzare la sua qualità di vita.

Trial terapeutici
AcetylcysteineAmantadineArmiclomorB1 ricombinant interferonBaclofenBCAABDNFCelecoxibCoenzymeQ10CreatineCyclosporineDextrometorphanGabapentinGangliosidesGlatiramerGlutathioneGuanidineIGF1IndinavirInosiplex
IsoprinosineLamotrigineLevamisoleLithium carbonateL-threonineMethionineMethylcobalaminMinocyclineNimodipinePentoxiphyllinePhysostigmineRecombinant clilary nerve growth factorRiluzoleSelegilineTCH346ThalidomideThyrotropineTiloroneTopiramate
Transfer factorValproic acidVitamin EXaliprodenErithropoietin

Problems about trial:• Rare disorder• Correct doses for neuroprotection are difficult to identify
• Outcomes are clinical (no biologic marker exist)
• Progressive weakness can lead to missing data

Targets:• Muscle proteins• Ways to stabilize energy expenditure
• Cell replacement therapies
• Silecing abnormal genes



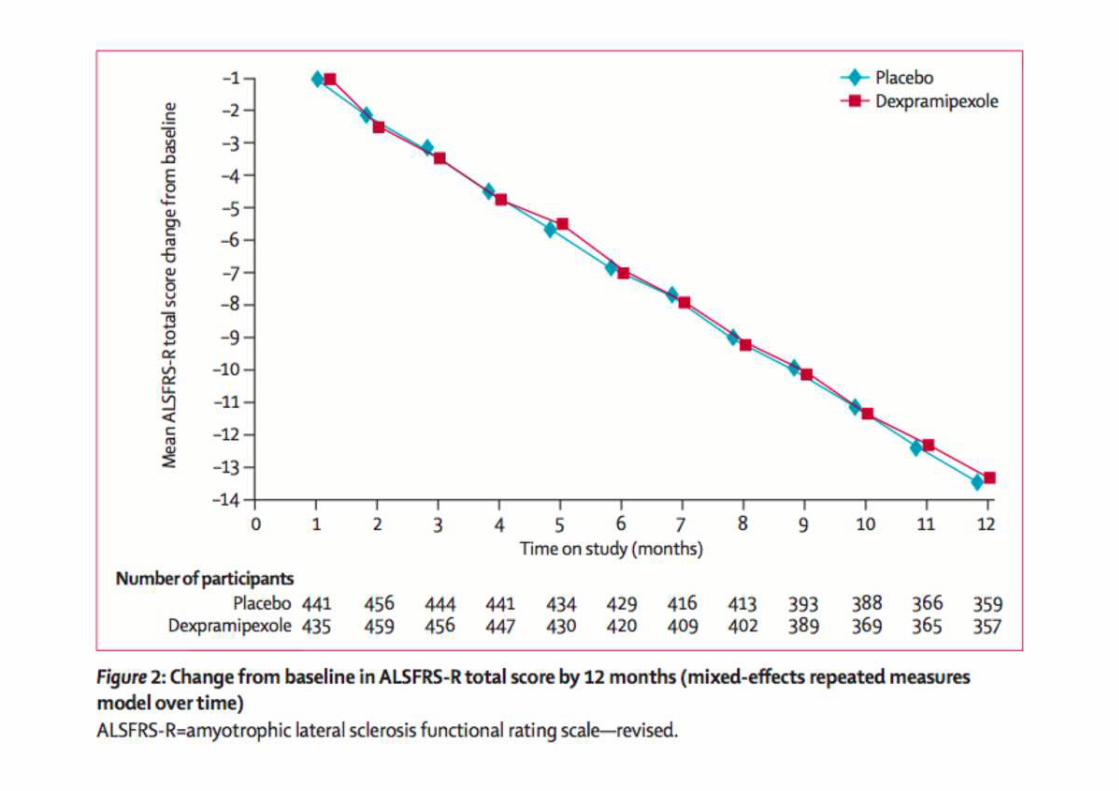
Trial terapeutici: dexpramipexolo

Stem cell therapy
Systemic
Local
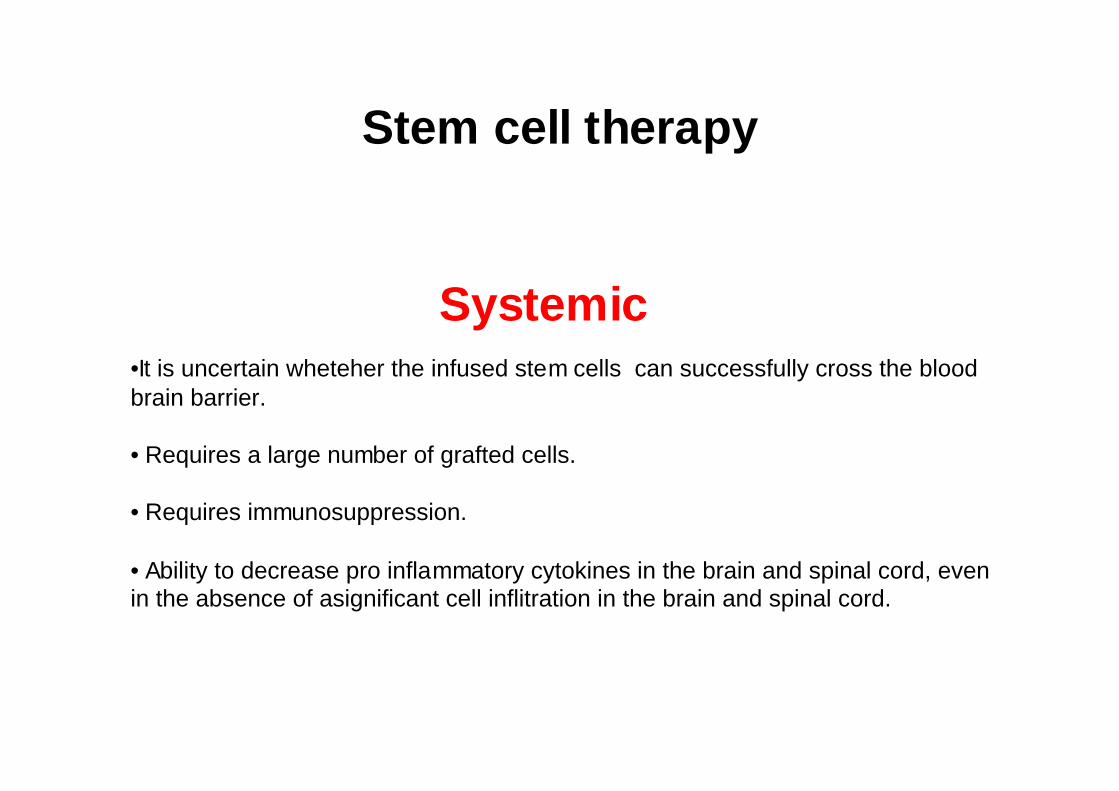
Stem cell therapy
Systemic•It is uncertain wheteher the infused stem cells can successfully cross the blood brain barrier.
• Requires a large number of grafted cells.
• Requires immunosuppression.
• Ability to decrease pro inflammatory cytokines in the brain and spinal cord, even in the absence of asignificant cell inflitration in the brain and spinal cord.

Stem cell therapy
Local• Multiple injections of stem cells along the entire length of the spinal cord.
• Precise migration of grafted cells to the desired destination.
• Risk of spinal cord injury.
• Immunosuppression not required.
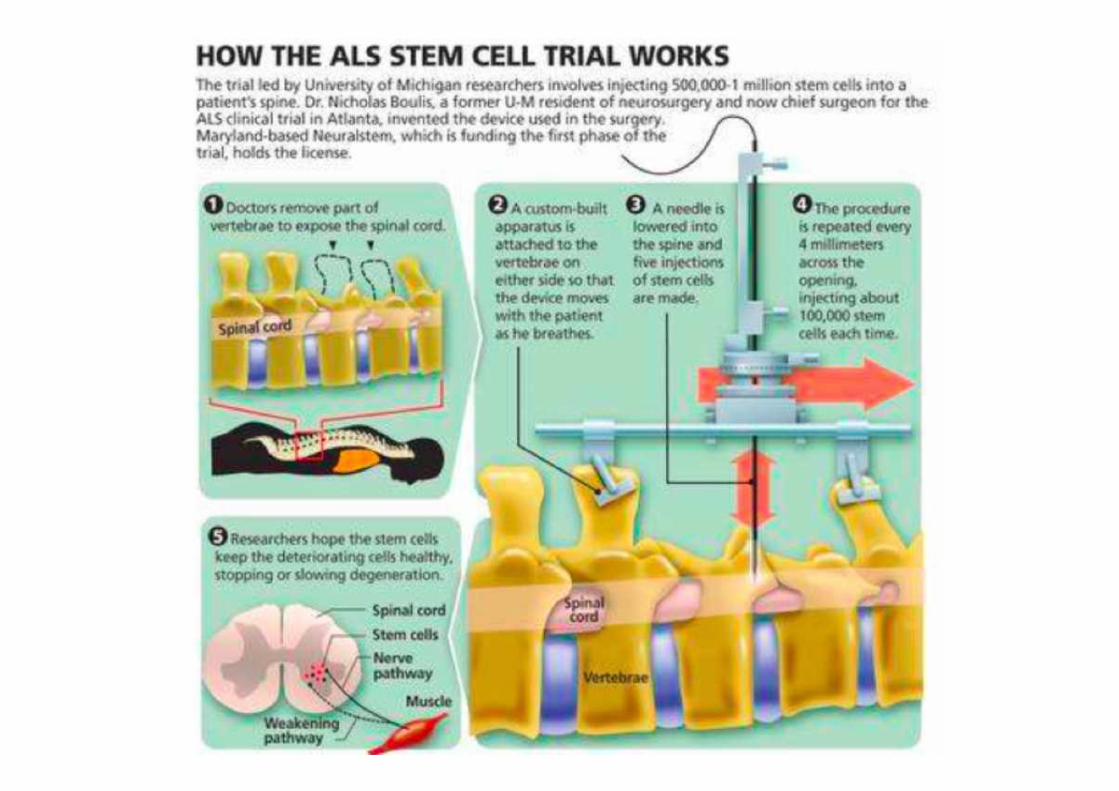

Stem cell therapy
Local• Stem cells can be injected into the subarachnoid space or the lateral ventricles
• Bypass the blood braim barrier.
• Reduces the risk of CNS injury.

Stem cell therapy… clinical trials
•.


Caso clinico 1 V.S.
• Pz uomo di 38 anni.
• Anamnesi patologica remota non significativa.
• Non assume farmaci.• Non fuma.
• Non storia di esposizione professionale a tossici.• Impiegato.
• Grande sportivo (nuoto, sci alpinismo, corsa).

Caso clinico 1
ANAMNESI PATOLOGICA PROSSIMA
Nel mese di Ottobre 2012 inizia a lamentare “movimenti muscolari involontari” al cingolo scapolare sn.
I “guizzi”muscolari si diffondono ai 4 arti.Il paziente a Novembre 2012 esegue una RMN
encefalo e midollo su indicazione del MMG: negativa.
I sintomi persistono senza calo di forza.

Caso clinico 1 VISITA NEUROLOGICA (APRILE 2013)
• Vigile, orientato, collaborante.
• Settore cranico indenne: bene i MOE,MOI, non deficit di VII, nondisartria, non disfagia, non disfonia. Lingua normo mobile.
• Non deficit in Barrè e Mingazzini ed alle prove segmentarie.
• Ipertrofia generale delle masse muscolari.
• Fascicolazioni diffuse evidenti ai muscoli degli arti e del tronco.
• Riflessi: vivaci (4+) ai 4 arti.
• Non Babinski.
• Non deficit di sensibilità.
• Deambulazione con lievi note pareto-spastiche.
• Non deficit sfinterici.

Caso clinico 1 VISITA NEUROLOGICA (APRILE 2013)
• Vigile, orientato, collaborante.
• Settore cranico indenne: bene i MOE,MOI, non deficit di VII, nondisartria, non disfagia, non disfonia. Lingua normo mobile.
• Non deficit in Barrè e Mingazzini ed alle prove segmentarie.
• Ipertrofia generale delle masse muscolari.
• Fascicolazioni diffuse evidenti ai muscoli degli arti e del tronco.
• Riflessi: vivaci (4+) ai 4 arti.
• Non Babinski.
• Non deficit di sensibilità.
• Deambulazione con lievi note pareto-spastiche.
• Non deficit sfinterici.
II motoneurone

Caso clinico 1 VISITA NEUROLOGICA (APRILE 2013)
• Vigile, orientato, collaborante.
• Settore cranico indenne: bene i MOE,MOI, non deficit di VII, nondisartria, non disfagia, non disfonia. Lingua normo mobile.
• Non deficit in Barrè e Mingazzini ed alle prove segmentarie.
• Ipertrofia generale delle masse muscolari.
• Fascicolazioni diffuse evidenti ai muscoli degli arti e del tronco.
• Riflessi: vivaci (4+) ai 4 arti.
• Non Babinski.
• Non deficit di sensibilità.
• Deambulazione con lievi note pareto-spastiche.
• Non deficit sfinterici.
I motoneurone

Caso clinico 1
QUALI IPOTESI DIAGNOSTICHE?
1 Sclerosi Laterale Amiotrofica (fascicolazioni…)
2 Patologia del SNC (iperreflessia, lieve spasticità)
3 Patologia muscolare primitiva (ipertrofia masse muscolari)

Caso clinico 1
QUALI ALTRI ESAMI FARESTE?
COME PROCEDERE NELLE INDAGINI?

Caso clinico 1
Luglio 2013: il paziente viene sottoposto ad EMG.
Elettroneurografia:- Normali parametri di conduzione dei nervi
sensitivi.
- Generale riduzione di ampiezza di tutt i inervi motori esplorati (sofferenza assonale).

Caso clinico 1
Elettromiografia:Con agoelettrodo vengono esaminati i muscoli TA,
gastrocnemio, estensore comune delle dita, deltoide, I°interosseo, linguale e mentoniero.
Riscontro di fascicolazioni sub continue, fibrillazione e PSW. Tracciato sub interferenziale-ridotto nello sforzo volontario massimale.

Caso clinico 1
Conclusioni:Segni di sofferenza neurogena acuta e cronica in
tutti idistretti muscolari esaminati (bulbare, cervicale e lombosacrale).
Il dato è indicativo di patologia del II motoneurone.

Caso clinico 1
Il paziente viene sottoposto a studio dei POTENZIALI EVOCATI MOTORI.
Conclusioni : Lieve aumento del tempo di conduzione motorio centrale per i 4 arti.

Caso clinico 1
Il paziente in data 25.05.2013 viene nuovamente sottoposto a RMN cerebrale.
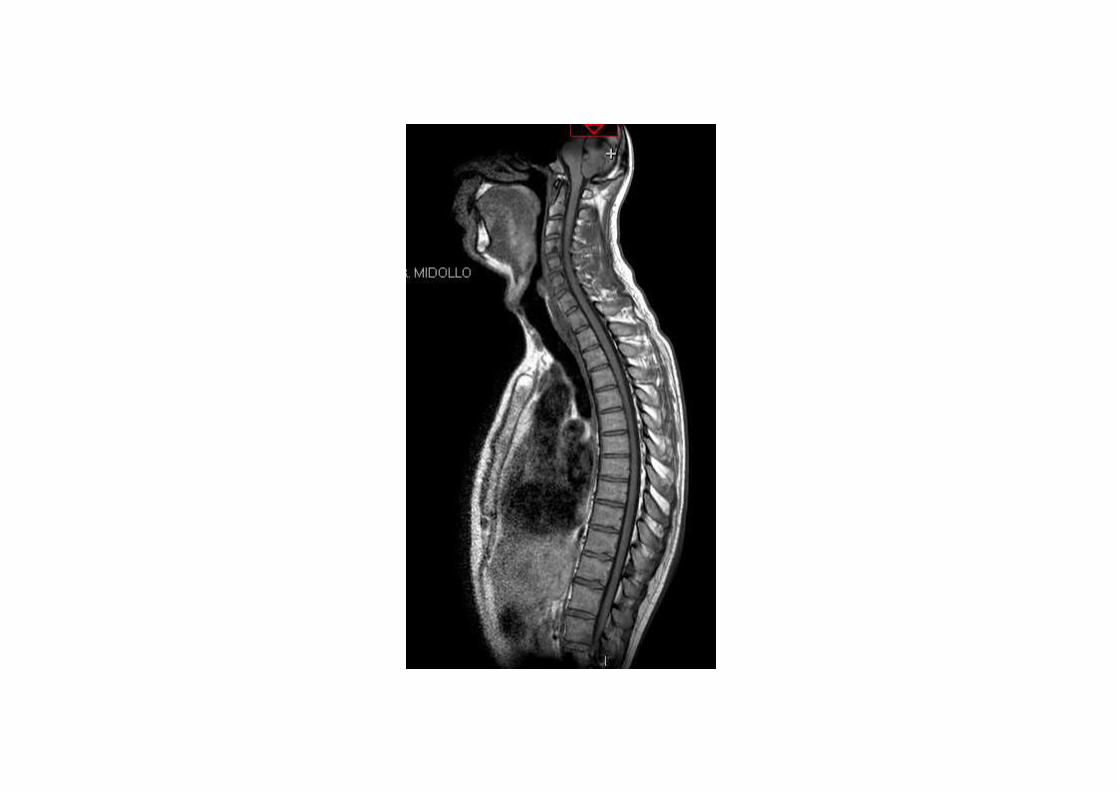

Caso clinico 1
Altri esami effettuati:
• Studio liquorale: nella norma.• Esami ematici: ndr. Autoimmunità e markers
neoplastici negativi. Incremento CPK (400U/I).

Caso clinico 1
Controllo clinico del 24.06.2013:
• ROT vivaci (4+).• Trofismo muscolare e forza elementare ben
conservata.
• Persistono fascicolazioni diffuse a tutti i distretti muscolari esclusa la lingua.
• Non Babinski.

Caso clinico 1
Controllo clinico del 31.12.2013:
• ROT vivaci (4+).• Trofismo muscolare e forza elementare ben
conservata ad eccezione di lieve atrofia del muscolo I interosseo di sinistra con ipostenia nell’afferramento.
• Persistono fascicolazioni diffuse a tutti i distretti muscolari esclusa la lingua.
• Babinski +.
• Riflesso masseterino vivace.
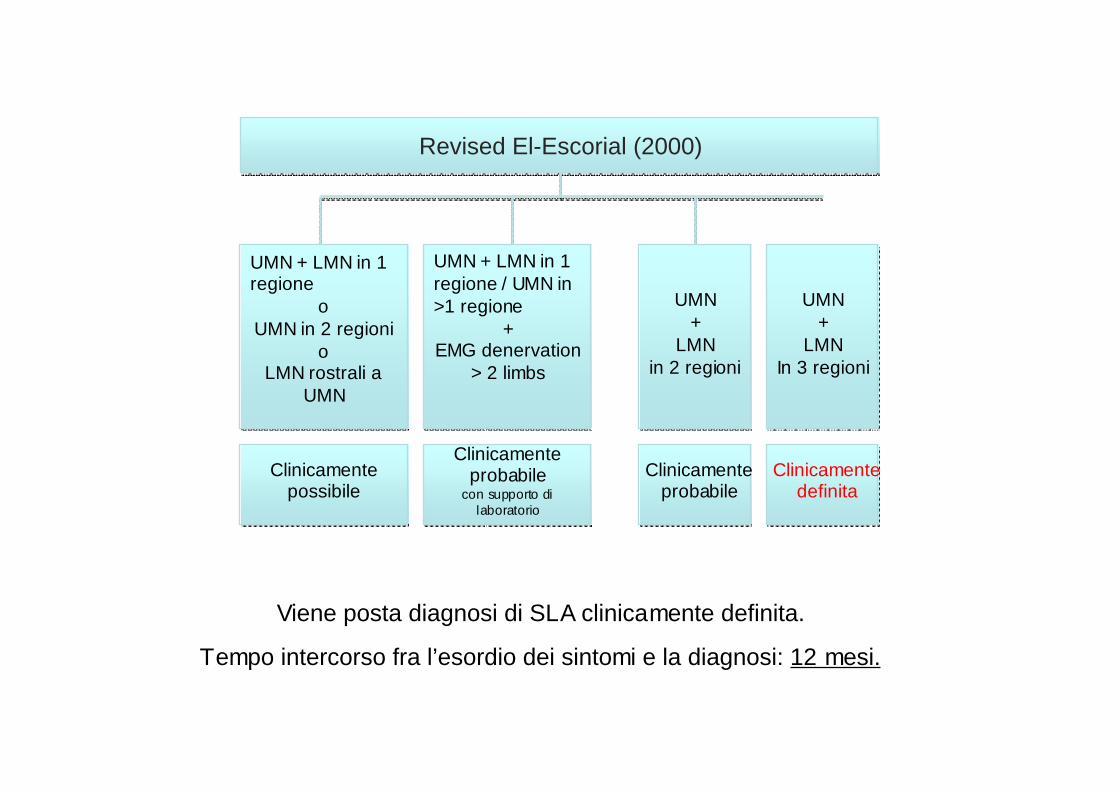
Revised El-Escorial (2000)Revised El-Escorial (2000)
Clinicamente possibile
Clinicamente probabile
con supporto di laboratorio
Clinicamente probabile
Clinicamente definita
UMN + LMN in 1 regione
oUMN in 2 regioni
oLMN rostrali a
UMN
UMN + LMN in 1 regione / UMN in >1 regione
+EMG denervation
> 2 limbs
UMN +
LMN in 2 regioni
UMN +
LMN In 3 regioni
Viene posta diagnosi di SLA clinicamente definita.
Tempo intercorso fra l’esordio dei sintomi e la diagnosi: 12 mesi.

Caso clinico 2 L.R.
• Pz donna di 45 anni.
• Anamnesi patologica remota: ipotiroidismo subclinico.
• Assume: Eutirox 50.
• Non fuma.• Non storia di esposizione professionale a tossici.

Caso clinico 2
ANAMNESI PATOLOGICA PROSSIMA
Novembre 2009: la paziente giunge a visita per disturbo della parola esordito a Luglio. Il disturbo è descritto come impaccio ad articolare le parole, accentuato da fattori emotivi o se parla a lungo.
Inoltre lamenta saltuaria disfagia e faticabilitàmasticatoria (non riesce più a mangiare il chewing gum).

Caso clinico 2
EON: vigile,orientata,collaborante. Parola lievemente abburrattata. Non deficit di MOE, MOI, non deficit dei nervi cranici V,VII, XII. In Barrè slivella senza pronare con l’arto superiore di destra, in Mingazzini slivellamento a dx. ROT vivaci.Non deficit alle prove I-N e C-G. Non deficit sensitivi. Non Babinski.

Caso clinico 2
QUALI IPOTESI DIAGNOSTICHE FARESTE?
patologia cerebrovascolare
SLA bulbare
miastenia gravis bulbare
Altre???

Caso clinico 2
Gennaio 2010: la paziente viene sottoposta a RMN encefalo.

Caso clinico 2
Gennaio 2010: viene eseguita una EMG. L’esame documenta fascicolazioni a carico dei muscoli bulbari (lingua, massetere, mentale), di alcuni muscoli del distretto cervicale e lombare.

Caso clinico 2
Febbraio 2010:
EON: vigile,orientata,collaborante. Parola abburrattata. Non deficit di MOE, MOI, non deficit dei nervi cranici V,VII. Fascicolazioni linguali e lingua lievemente improntata. Ipotrofia ed ipostenia dei muscoli I interosseo bilateralmente. Fascicolazioni evidenti ai muscoli tricipite bilaterale, deltoide e ai muscoli delle cosce.ROT vivaci ai 4 arti. Non deficit alle prove I-N e C-G. Non deficit sensitivi. Babinski +. Riflesso masseterino vivace.
I motoneurone

Caso clinico 2
Febbraio 2010:
EON: vigile,orientata,collaborante. Parola abburrattata. Non deficit di MOE, MOI, non deficit dei nervi cranici V,VII. Fascicolazioni linguali e lingua lievemente improntata. Ipotrofia ed ipostenia dei muscoli I interosseo bilateralmente. Fascicolazioni evidenti ai muscoli tricipite bilaterale, deltoide e ai muscoli delle cosce.ROT vivaci ai 4 arti. Non deficit alle prove I-N e C-G. Non deficit sensitivi. Babinski +. Riflesso masseterino vivace. II motoneurone

Revised El-Escorial (2000)Revised El-Escorial (2000)
Clinicamente possibile
Clinicamente probabile
con supporto di laboratorio
Clinicamente probabile
Clinicamente definita
UMN + LMN in 1 regione
oUMN in 2 regioni
oLMN rostrali a
UMN
UMN + LMN in 1 regione / UMN in >1 regione
+EMG denervation
> 2 limbs
UMN +
LMN in 2 regioni
UMN +
LMN In 3 regioni
Viene posta diagnosi di SLA clinicamente definita.
Tempo intercorso fra l’esordio dei sintomi e la diagnosi: 8 mesi.

• A dicembre 2010 la paziente inizia la Ventilazione meccanica non invasiva per iniziale insufficienza respiratoria.
• Nel corso del 2010-2011 sviluppa progressiva ipostenia ed ipotrofia ai 4 arti, non è più in grado di muovere nulla.
• La parola non è più comprensibile.
• 24.10.2012: dopo un accesso al PS per insufficienza respiratoria acuta ed evidenza all’rx torace di addensamento basale, PREVIO CONSENSO INFORMATO della paziente, viene sottoposta a tracheotomia in emergenza.
• Dopo un breve ricovero in Rianimazione viene trasferita in Riabilitazione Pneumologica a Correggio dove resta un mese, qui viene sottoposta anche a PEG per impossibilità ad alimentarsi.
• La famiglia viene addestrata all’assistenza e preparata al rientro della paziente al domicilio.
•2014: la paziente vive al domicilio. Assistenza del SID.

Caso clinico 3 G.P.
• Pz uomo di 73 anni.
• Cardiopatia ischemica (IMA 1993 e 2005).
• Diabete tipo 2.• Ateromasia TSA.
• Fumatore.• Attività lavorativa: ex impiegato (pensionato).
Caso clinico 3
ANAMNESI PATOLOGICA PROSSIMA
• Febbraio 2013: esordio di ipostenia alla mano sinistra (difficoltà nell’opposizione pollice-indice) cin sviluppo di ipotrofia del m. I°intersseo.

Caso clinico 3
ANAMNESI PATOLOGICA PROSSIMA
• Nell’arco di 2 mesi il paziente nota dimagrimento della massa muscolare con calo ponderale di circa 7 Kg, astenia generale, fascicolazioni ai muscoli di gambe e braccia, crampi ai polpacci.

Caso clinico 3
Ottobre 2013: Visita neurologica.
EON: ipotrofia dell’emilingua senza fascicolazioni evidenti. Ipotrofia dei muscoli del cingolo scapolare, bicipite, tricipite brachiale, I°interosseo, APB sn>dx. Ipotrofia anche del muscolo quadricipite femorale con fascicolazioni. ROT vivaci AASS. Risposta scorretta alla stimolazione cutanea plantare. Stazione eretta e marcia nei limiti.

Caso clinico 3
Il paziente viene ricoverato.
• RMN cervicale: cervico atrosi di modesta entità.
• Rachicentesi: lieve rialzo della quota di proteine. IEF n.n.
• EMG: dati NF compatibili con segni di sofferenza del II motoneurone.
• Potenziali Evocati Motori:ritardo di conduzione cordonale spinale bilaterale.

Caso clinico 3
• RMN encefalo: segni di sofferenza vascolare cronica.
• Trattografia: “lo studio trattografico mostra ridotto valore medio dell’anisoptropia frazionale a carico del fascio piramidale e della via cortico pontina del lato di destra rispetto al controlaterale.”

Caso clinico 3
• Sulla base dei segni di coinvolgimento del II m.n (lingua, arti superiori ed inferiori), visti i dati strumentali (EMG, PEM e RMN con trattografia)e quelli clinici (ROT vivaci e SCP scorretto come segni di I m.n, atrofia muscolare come segno di II m.n) viene posta diagnosi di SLA.
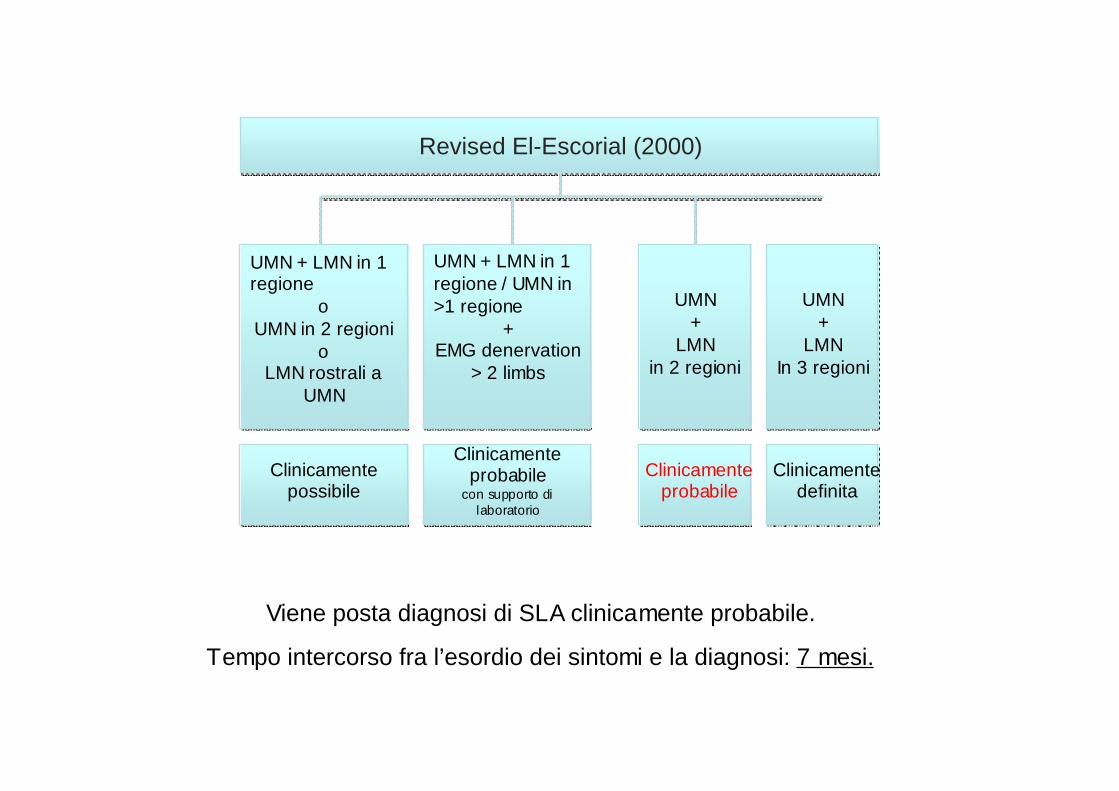
Revised El-Escorial (2000)Revised El-Escorial (2000)
Clinicamente possibile
Clinicamente probabile
con supporto di laboratorio
Clinicamente probabile
Clinicamente definita
UMN + LMN in 1 regione
oUMN in 2 regioni
oLMN rostrali a
UMN
UMN + LMN in 1 regione / UMN in >1 regione
+EMG denervation
> 2 limbs
UMN +
LMN in 2 regioni
UMN +
LMN In 3 regioni
Viene posta diagnosi di SLA clinicamente probabile.
Tempo intercorso fra l’esordio dei sintomi e la diagnosi: 7 mesi.

Caso clinico 4 R.A.
• Pz uomo di 73 aa.
• Nel 2004 inizia a lamentare difficoltà nella deambulazione per senso di cedimento dell’AI dx. Astenia generalizzata.
• L’EON ad una prima visita neurologica ènegativo.

Caso clinico 4
• Nel 2005 un’ EMG rileva segni di sofferenza neurogena cronica compatibili con malattia del motoneurone.
• Una RMN cervicale descrive un’ernia discale C6-C7 su un quadro di artrosi.
• In regime di DH esegue rachicentesi (n.n.) e ANA test, ANCA, markers neoplastici, IgM ed IgG anti Borrelia, Crioglobulinemia: n.n.

Caso clinico 4
• Dal 2005 al 2006 il pz viene seguito con EMG periodiche che risultavano invariate.
• Ex adiuvantibus è stato trattato con Ig ev.

Caso clinico 4
• Nel 2006 all’ipostenia riferita si aggiunge una lieve disfonia e e ristagno di saliva.
• Il controllo EMG documenta fascicolazioni linguali.

Caso clinico 4
• Dal 2007 al 2008 esegue altre EMG, invariate.
• L’EON è negativo.
• Test genetico per M.di Kennedy: negativo.• Potenziali Evocati Motori: alterazione della via
motoria centrale ai 4 arti.

Caso clinico 4
• Dal 2009 al 2012 il paziente non riferisce variazioni cliniche importanti eccetto l’urgenza minzionale (inizialmente attribuito al quadro di ipertrofia prostatica).

Caso clinico 4
• Dal 2012 vi è un rapido peggioramento dell’ipostenia con frequenti cadute.
• Ottobre 2013: il pz viene ricoverato per accertamenti.
• EON: Nulla al settore cranico e al distretto degli arti superiori. Ipertono spastico dell’AI dx. Deambulazione falciante a destra. ROT piùvivaci agli arti di destra con clono achilleo a dx.
I motoneurone

Caso clinico 4
Il paziente viene sottoposto a trattografia che mostra segnale compatibile con possibile coinvolgimento primario della via cortico spinale (sebbene i dati non assumano un valore di specificità).

Caso clinico 4
COSA POSSIAMO CONCLUDERE?
QUALE DIAGNOSI?
QUALI DIAGNOSI DIFFERENZIALI?

Caso clinico 4
• Alla luce del coinvolgimento della via piramidale, dimostrato a livello clinico e strumentale mediante RMN e PEM si conclude per Sclerosi Laterale Primaria (SLA primaria, a coinvolgimento esclusivo del I°mn).

Arch Neurol vol 64, Feb2007
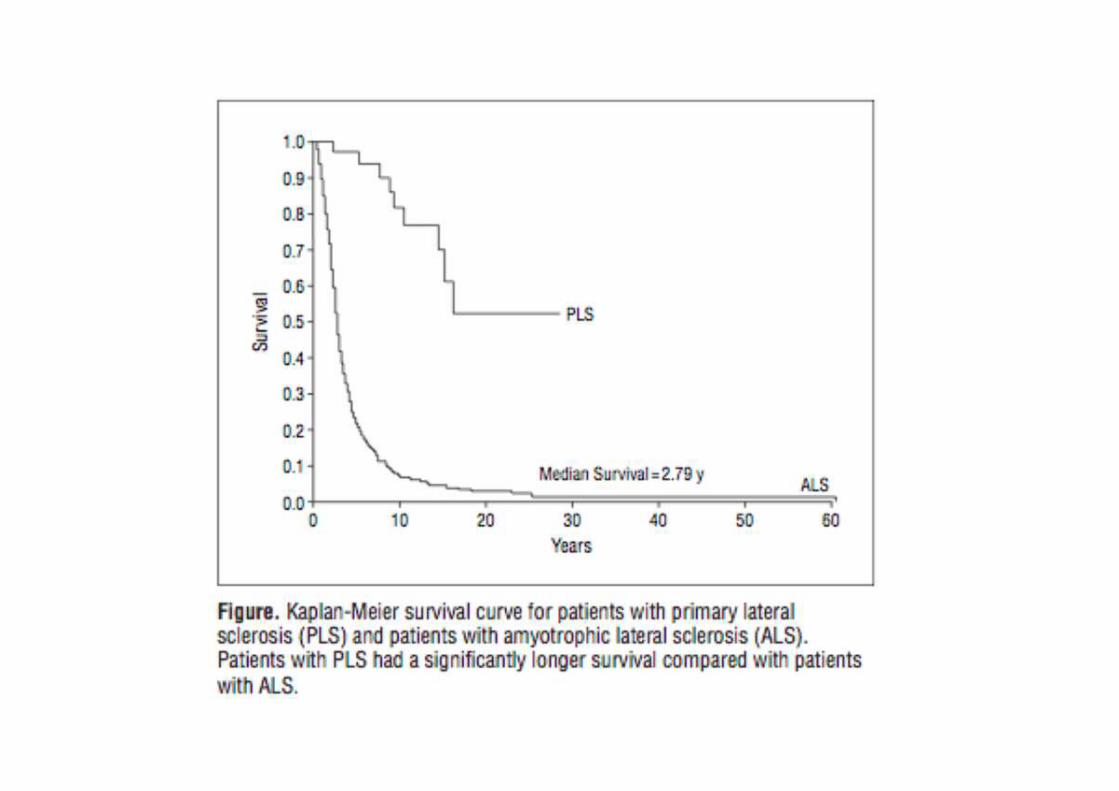

I FENOTIPI CLINICI NELLA SLA:I CONFINI SI AMPLIANO
PLSPrimary Lateral Sclerosis
Classical ALS Bulba r Flail Arm Flail LegPMAPrimary Muscular Atrophy
Flail Arm: brachial amyotrophic diplegia.Flail Leg : pseudopolyneuritic variant

Flail arm variant

Flail leg variant
Forma pseudopolinevritica
Debolezza distale AAII
Coinvolgimento asimmetrico AAI
ROT asseti
Lenta progressione
Lievi segni di coinvolgimento di I°mn

Caso clinico : ALS mimiking syndrome
• Pz uomo di 56 anni.• Iperteso.
• Anamnesi patologica remota per il resto non significativa.
• Non assume farmaci.• Non fuma.• Non storia di esposizione professionale a
tossici.

• Il paziente giunge alla nostra attenzione per progressivo sviluppo nell’arco di un anno di ipostenia ed ipotrofia bilaterale della muscolatura degli AASS (spalle e braccia).
• Non dolore.• Negativo un precedente studio RMN
encefalo-cervicale effettuato un anno prima.

• EON: vigile, collaborante. Non deficit dei nervi cranici. Non segni bulbari (disfonia, disartria, ipoestenia facciale o linguale). Non deficit di motilità oculare. In Barrè slivella bilateralmente per ipostenia dei mm bicipite e deltoide, meglio conservata la forza distale (interossei). Atrofia dei mm bicipite, tricipite, deltoide. Visibili fascicolazioni ai mm tricipite. ROT vivaci AASS. Non deficit stenici agli AAII ma ROT rotulei vivaci. SCP: in flessione bilaterale. Non deficit sensitivi o di coordinazione.

• Liquor: n.n.
• EMG: normali parametri di conduzione dei nervi esaminati. All’esame ad ago rilievo di fascicolazioni ai muscoli bicipite e tricipite bilateralmente ma non fibrillazione né PSW.
• Biopsia del m.bicipite: segni di atrofia muscolare.
• TMS: prolungamento del tempo di conduzione centrale sia per gli AASS che per gli AAII.

• Sulla base del quadro clinico e della negativitàdella precedente RM viene formulata una diagnosi di SLA possibile secondo i criteri di El Escorial.
Revised El-Escorial (2000)Revised El-Escorial (2000)
Clinicamente possibile
Clinicamente probabile
con supporto di laboratorio
Clinicamente probabile
Clinicamente definita
UMN + LMN in 1 regione
oUMN in 2 regioni
oLMN rostrali a
UMN
UMN + LMN in 1 regione / UMN in >1 regione
+EMG denervation
> 2 limbs
UMN +
LMN in 2 regioni
UMN +
LMN In 3 regioni
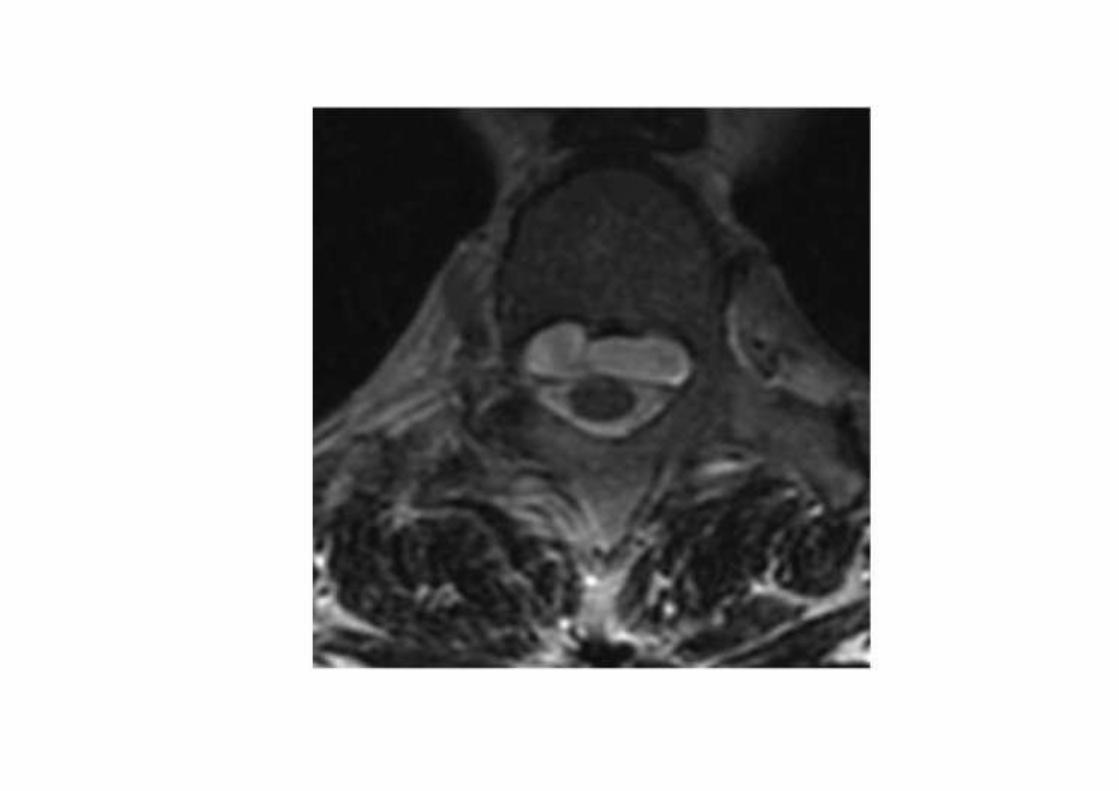
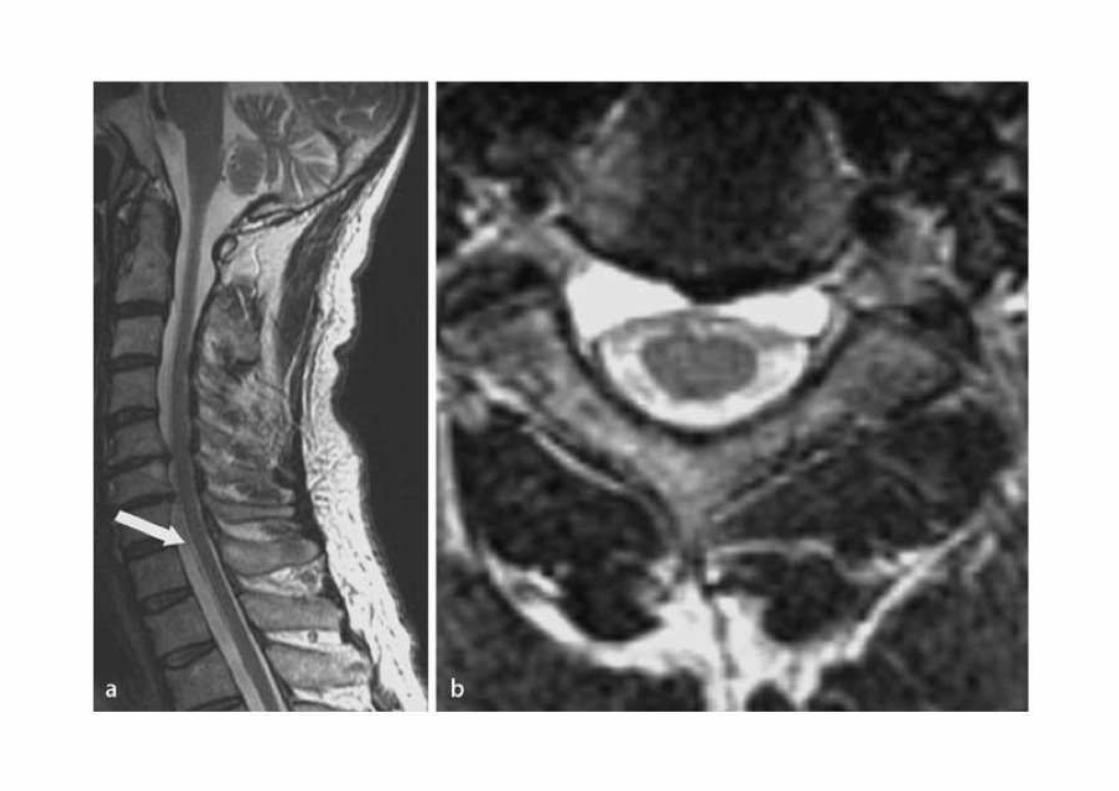

• Il paziente rifiutò l’intervento NCH.• Il follow up neuroradiologico a 6 e 12 mesi
rivelò stazionarietà.• Stazionarietà clinica a 12 mesi con sintomi
controllati con gabapentin e FANS.


















