
Ipogonadismo e infertilit
5
dicembre Vol. 13, n° 6 Endocrinologia - Dipartimento di Scienze Cardiotoraciche e Respiratorie, Seconda Università degli Studi di Napoli, Napoli Corrispondenza: Prof. A.A. Sinisi, Endocrinologia - Dipartimento di Scienze Cardiotoraciche e Respiratorie, Seconda Università di Napoli, Via Pansini 5, 80121 Napoli. E-mail: [email protected] Ipogonadismo e infertilità in ragazzi con sindromi genetiche rare Proposto da Antonio Agostino Sinisi I pogonadismo ed infertilità possono essere presenti in ragazzi con sindromi genetiche rare dominate da malformazioni somatiche o alterazioni siste- miche. I disordini riproduttivi sono dovuti a difetti testicolari primitivi (sindrome di Klinefelter, del maschio XX, di Noonan, difetto d’azione degli androgeni, iperplasia surrenale congenita), secondari (sindrome di Kallmann, deficit ipofisario multiplo, displasia setto ottica, sindrome di Bardet-Biedl) o misti (sindrome di Prader-Willi, distrofia miotonica tipo 2, atassia cerebellare, sindrome di Wolfram), oppure ad alterazioni post-testi- colari (s. Denys-Drash, Frasier, malattia policistica renale, discinesia cilia- re primitiva, sordità-infertilità). L’ipogonadismo è in genere diagnosticato in età adolescenziale. La diagnosi di ipogonadismo sindromico e del difetto genetico è importante per la definizione eziologica e la prevenzione. © 2012, Editrice Kurtis L’infertilità maschile è una condi- zione eterogenea, che nel 15% dei casi ha un’eziologia genetica. Sia alterazioni cromosomiche (numeri- che, strutturali o microdelezioni) sia disordini monogenici o poligenici possono determinare una condizione di ipogonadismo e/o infertilità (1). In rari casi l’ipogonadismo e l’infer- tilità possono essere aspetti di un quadro sindromico complesso, nel quale le alterazioni extra-riprodutti- ve, come malformazioni somatiche o disordini sistemici (neurologici, car- diovascolari, polmonari, renali, ossei), sono prevalenti e portano il soggetto all’osservazione medica già in età pediatrica. Nei ragazzi affetti da queste sindromi le alterazioni riproduttive (ambiguità dei genitali, criptorchidismo, ritardo puberale, ipogonadismo, anomalie deferenzia- li, alterazioni biochimico-funzionali degli spermatozoi) possono essere presenti singolarmente o associarsi tra di loro e sono dovute a: 1) difetti primitivi testicolari, 2) difetti della sintesi o del rilascio delle gonado- tropine, 3) alterazioni post-testicola- ri uro-genitali o anomalie biomole- colari dei gameti. SINDROMI CON DIFETTI PRIMITIVI TESTICOLARI Anomalie dei genitali (criptorchi- dismo, ipotrofia testicolare, micrope- ne, ambiguità dei genitali esterni, persistenza di derivati mulleriani come utero o tube rudimentali), alte- razioni somatiche (statura, ginecoma- stia ecc.), viscerali (cardiache, renali, neurologiche ecc.) e disturbi psico- comportamentali possono essere pre- senti in sindromi associate ad aberra- zioni dei cromosomi sessuali o ad alterazioni di geni presenti sui gono- somi o sugli autosomi (Tabelle 1 e 2). La sindrome di Klinefelter è un’a- nomalia cromosomica frequente negli uomini infertili (prevalenza di 1:1.000 in età adulta, fino a 1:100 negli azoospermici). In età infantile Antonio Agostino Sinisi, Iolanda Cioffi, Daniela Visconti, Giuseppe Bellastella, Vincenzo Palumbo le manifestazioni cliniche sono sfu- mate, mentre l’ipogonadismo iper- gonadotropo appare negli stadi intermedi o avanzati della pubertà. Le altre sindromi da anomalie cro- mosomiche sono molto più rare e talora sono diagnosticate nell’infan- zia per l’ambiguità dei genitali. La sindrome del maschio XX o disordine testicolare della differen- ziazione sessuale (testicular-DSD) ha una prevalenza di 1:20.000 ed è caratterizzata da un fenotipo maschi- le con cariotipo 46,XX, bassa statu- ra, azoospermia e ipogonadismo ipergonadotropo. Il 90% dei casi ha il gene SRY (sex-determining region on the Y) traslocato sul cromosoma X o su un autosoma (SRY-positivi), geni- tali maschili normali, testicoli pic- coli e azoospermia. I casi senza SRY (soggetti SRY-negativi) hanno gine- comastia, genitali ambigui e talora gonadi con tessuto testicolare e ova- rico (ovotesticular-DSD) (2). La diffe- renziazione in senso maschile avvie- ne per effetto dell’azione del prodot- to di SRY, mentre, in assenza di SRY, è dovuta all’iper-espressione della cascata regolata dai geni SOX (Sry-related HMG-box). La secrezione di testosterone può essere carente fin dall’infanzia o ridursi con la pubertà con comparsa di ipogonadismo iper- gonadotropo post-puberale. L’azoo- spermia nel maschio XX è dovuta all’assenza della regione AZF del cromosoma Y. La sindrome di Noonan (NS) è un disordine autosomico dominante caratterizzato clinicamente da bassa statura, dismorfismi facciali, pteri- gio, difetti cardiaci congeniti, ano- malie scheletriche ed ematologiche e 259
-
Upload
daniela-visconti -
Category
Documents
-
view
219 -
download
3
Transcript of Ipogonadismo e infertilit

dicembre Vol. 13, n° 6
Endocrinologia - Dipartimento di Scienze Cardiotoraciche e Respiratorie,Seconda Università degli Studi di Napoli, Napoli
Corrispondenza: Prof. A.A. Sinisi, Endocrinologia - Dipartimento di Scienze Cardiotoraciche e Respiratorie, SecondaUniversità di Napoli, Via Pansini 5, 80121 Napoli. E-mail: [email protected]
Ipogonadismo e infertilitàin ragazzi con sindromi
genetiche rareProposto da Antonio Agostino Sinisi
Ipogonadismo ed infertilità possono essere presenti in ragazzi con sindromigenetiche rare dominate da malformazioni somatiche o alterazioni siste-
miche. I disordini riproduttivi sono dovuti a difetti testicolari primitivi(sindrome di Klinefelter, del maschio XX, di Noonan, difetto d’azione degliandrogeni, iperplasia surrenale congenita), secondari (sindrome diKallmann, deficit ipofisario multiplo, displasia setto ottica, sindrome diBardet-Biedl) o misti (sindrome di Prader-Willi, distrofia miotonica tipo 2,atassia cerebellare, sindrome di Wolfram), oppure ad alterazioni post-testi-colari (s. Denys-Drash, Frasier, malattia policistica renale, discinesia cilia-re primitiva, sordità-infertilità). L’ipogonadismo è in genere diagnosticatoin età adolescenziale. La diagnosi di ipogonadismo sindromico e del difettogenetico è importante per la definizione eziologica e la prevenzione.©2012, Editrice Kurtis
L’infertilità maschile è una condi-zione eterogenea, che nel 15% deicasi ha un’eziologia genetica. Siaalterazioni cromosomiche (numeri-che, strutturali o microdelezioni) siadisordini monogenici o poligenicipossono determinare una condizionedi ipogonadismo e/o infertilità (1).In rari casi l’ipogonadismo e l’infer-tilità possono essere aspetti di unquadro sindromico complesso, nelquale le alterazioni extra-riprodutti-ve, come malformazioni somatiche odisordini sistemici (neurologici, car-diovascolari, polmonari, renali,ossei), sono prevalenti e portano ilsoggetto all’osservazione medica giàin età pediatrica. Nei ragazzi affettida queste sindromi le alterazioniriproduttive (ambiguità dei genitali,criptorchidismo, ritardo puberale,ipogonadismo, anomalie deferenzia-li, alterazioni biochimico-funzionalidegli spermatozoi) possono esserepresenti singolarmente o associarsitra di loro e sono dovute a: 1) difettiprimitivi testicolari, 2) difetti dellasintesi o del rilascio delle gonado-tropine, 3) alterazioni post-testicola-ri uro-genitali o anomalie biomole-colari dei gameti.
SINDROMI CON DIFETTI PRIMITIVITESTICOLARI
Anomalie dei genitali (criptorchi-dismo, ipotrofia testicolare, micrope-ne, ambiguità dei genitali esterni,persistenza di derivati mullerianicome utero o tube rudimentali), alte-razioni somatiche (statura, ginecoma-stia ecc.), viscerali (cardiache, renali,neurologiche ecc.) e disturbi psico-comportamentali possono essere pre-senti in sindromi associate ad aberra-zioni dei cromosomi sessuali o adalterazioni di geni presenti sui gono-somi o sugli autosomi (Tabelle 1 e 2).
La sindrome di Klinefelter è un’a-nomalia cromosomica frequentenegli uomini infertili (prevalenza di1:1.000 in età adulta, fino a 1:100negli azoospermici). In età infantile
Antonio Agostino Sinisi, Iolanda Cioffi, Daniela Visconti,Giuseppe Bellastella, Vincenzo Palumbo
le manifestazioni cliniche sono sfu-mate, mentre l’ipogonadismo iper-gonadotropo appare negli stadiintermedi o avanzati della pubertà.
Le altre sindromi da anomalie cro-mosomiche sono molto più rare etalora sono diagnosticate nell’infan-zia per l’ambiguità dei genitali.
La sindrome del maschio XX odisordine testicolare della differen-ziazione sessuale (testicular-DSD) hauna prevalenza di 1:20.000 ed ècaratterizzata da un fenotipo maschi-le con cariotipo 46,XX, bassa statu-ra, azoospermia e ipogonadismoipergonadotropo. Il 90% dei casi hail gene SRY (sex-determining region onthe Y) traslocato sul cromosoma X osu un autosoma (SRY-positivi), geni-tali maschili normali, testicoli pic-coli e azoospermia. I casi senza SRY(soggetti SRY-negativi) hanno gine-
comastia, genitali ambigui e taloragonadi con tessuto testicolare e ova-rico (ovotesticular-DSD) (2). La diffe-renziazione in senso maschile avvie-ne per effetto dell’azione del prodot-to di SRY, mentre, in assenza diSRY, è dovuta all’iper-espressionedella cascata regolata dai geni SOX(Sry-related HMG-box). La secrezionedi testosterone può essere carente findall’infanzia o ridursi con la pubertàcon comparsa di ipogonadismo iper-gonadotropo post-puberale. L’azoo-spermia nel maschio XX è dovutaall’assenza della regione AZF delcromosoma Y.
La sindrome di Noonan (NS) è undisordine autosomico dominantecaratterizzato clinicamente da bassastatura, dismorfismi facciali, pteri-gio, difetti cardiaci congeniti, ano-malie scheletriche ed ematologiche e
259

260
Ipogonadismo maschile sindromico
difetti cognitivi variabili. Mutazionidi almeno nove geni (PTN11, SOS1,KRAS, NRAS, RAF1, BRAF,SHOC2, MEK1, CBL) possono darela NS o sindromi fenotipicamentecorrelate [sindrome di Leopard, diCostello, cardiofaciocutanea, NS-LAH (Noonan-like syndrome with looseanagen hair), neurofibromatosi-NS,di Legius]. Tutte queste condizionivengono raggruppate nella famigliadelle sindromi neurocardiofaciocuta-nee o delle RAS-patie, perché ricon-ducibili ad alterazioni della cascataRAS e si associano a un alto rischiodi sviluppare neoplasie. Oltre allabassa statura vi sono il criptorchidi-smo (in oltre il 50%) e il ritardopuberale. In età adulta è frequente ilriscontro di azoospermia o oligozoo-spermia, dovute a un’alterazione pri-mitiva testicolare disgenetica, checompromette la funzione sertoliana eleydigiana, ma anche al criptorchidi-smo (3).
Un difetto primitivo testicolare èpresente nel nanismo muliebre(MUL), una sindrome molto raradescritta nei finnici dovuta a muta-zione del gene TRIM37, che codifi-ca per una ubiquitin E3 ligase.L’ipogonadismo compare a metàpubertà con una riduzione progressi-va delle cellule germinali, del volu-me testicolare, dei livelli di testoste-rone e inibina B, e un paralleloaumento di LH e FSH. Gli adultipresentano azoospermia o oligozoo-spermia (4).
Le alterazioni della sintesi steroideadovute a difetti congeniti delle viebiosintetiche interessanti la gonadee/o il surrene (Tabella 2) hanno conun’ereditarietà di tipo autosomicorecessivo. Il difetto enzimatico puòdeterminare in epoca fetale un deficitdella sintesi del testosterone, permutazioni dei geni NR5A1 o SF1,STAR, CYP17, HSD3B2, HSD3B3,POR, oppure un eccesso di produzio-
ne di androgeni surrenalici (iperplasiasurrenale congenita, ISC) per muta-zione di CYP21A2 e CYP11A1. Neconsegue un’alterazione della diffe-renziazione sessuale del feto con effet-ti fenotipici diversi a seconda deldifetto, del sesso cromosomico e dellagonade. Nei soggetti geneticamentemaschi con difetti di biosintesi deltestosterone si ha ambiguità dei geni-tali e inversione del fenotipo sessuale,nei deficit surrenalici con aumentodegli androgeni si ha ipervirilizzazio-ne (macrogenitosomia, aumento dellapeluria corporea, precoce maturazioneossea, pubertà precoce e bassa statura).Gli uomini con ISC hanno frequente-mente alterazioni della spermatogene-si e aumento dei tumori testicolari daresidui surrenalici (5). Le alterazionitesticolari possono essere la conse-guenza dello stimolo cronicodell’ACTH elevato oppure della sop-pressione della secrezione delle gona-dotropine.
Sindrome Alterazione genetica Manifestazioni cliniche
S. Klinefelter Polisomia del cromosoma X Statura aumentata, proporzioni eunucoidi, ginecomastia,(47,XXY, 48,XXXY o altre polisomie X o mosaicismi) testicoli piccoli e duri, ipogonadismo ipergonadotropo,
azoospermia, oligozoospermia severa o criptozoospermia
S. maschio XX Traslocazione di SRY X-Y (SRY-positivi) Bassa statura, ipogonadismo ipergonadotropo10% SRY-negativi (overespressione SOX) ad insorgenza peripuberale, ipotrofia testicolare,
azoospermia (SRY-positivi o raramente SRY-negativi),ginecomastia, ambiguità dei genitali e ovotestis (SRY-negativi)
Disgenesia gonadica mista Mosaicismi XY/X0 o varianti Bassa statura, genitali ambigui, utero o tube rudimentali,gonadi asimmetriche ritenute con ipotrofia del testicolo o tumore(gonadoblastoma) da un lato e disgenesia controlaterale,ipogonadismo ipergonadotropo, utero rudimentale
S. maschio XYY e varianti Cromosoma Y sovrannumerario Ipogonadismo incostante, anomalie staturali, ossee e dentarie
S. Noonan e Noonan simili AD Bassa statura, anomalie cardiache e polmonari, anomalie somatiche,(S. neurocardiofaciocutanee o RAS-patie) Mutazioni di PTN11, SOS1, KRAS, NRAS, pterigio, ipogonadismo ipergonadotropo, criptorchidismo
RAF1,BRAF, SHOC2, MEK1, CBL
Displasia camptomelica AR Bassa statura, anomalie scheletriche, ipogonadismo ipergonadotropo,Mutazioni di SOX-9 residui testicolari, genitali femminilizzati
Persistenza di derivati mulleriani nel maschio AR Utero o altri derivati mulleriani, criptorchidismoMutazioni di AMH o AMHR
Nanismo muliebre (MUL) AR Disturbo di crescita prenataleMutazioni di TRIM37 Scafocefalia, sella a J, epatomegalia, displasia fibrosa,
pericardite restrittiva, ipogonadismo ipergonadotropoad esordio nel periodo medio-puberale
Tabella 1Difetti primitivi gonadici e alterazioni della differenziazione sessuale.
L’ambiguità dei genitali esternicaratterizza le sindromi da difetto del-l’azione androgena come l’insensibi-lità agli androgeni per mutazione delgene del recettore degli androgeni(AR), che si trasmette per via X-linked, o la resistenza agli androgeniper mutazione del gene della 5α-reduttasi 2 (SRD5A2), che ha eredita-rietà autosomica recessiva. Il fenotipopuò essere molto variabile. All’e-stremo dello spettro clinico vi puòessere solo l’ipospermatogenesi (6).
SINDROMI ASSOCIATE ADALTERAZIONI DELLA SINTESIE DEL RILASCIO DELLEGONADOTROPINE
Alcune sindromi geneticamentedeterminate presentano ipogonadi-smo ipogonadotropo, per alterazionecentrale della secrezione di GnRH odelle gonadotropine, associato adalterazioni sensoriali (anosmia, ipo-smia, sordità, cecità ai colori), cuta-nee (ittiosi), della linea mediana(palatoschisi, anomalie dentarie),neurologiche, renali, degli arti (sin-dattilia, polidattilia), ad altri difettiormonali (ipofisari, surrenalici), obe-sità o altre anomalie metaboliche.L’ipogonadismo ipogonadotropodetermina ritardo o assenza dello svi-luppo puberale o, più raramente, un
deficit gonadico a comparsa in etàadulta. La terapia sostitutiva congonadotropine ripristina la funzione
gonadica e la spermatogenesi (7). Inquesto gruppo (Tabella 3) rientranola sidrome di Kallmann e altre forme
261
ANTONIO AGOSTINO SINISI ET AL.
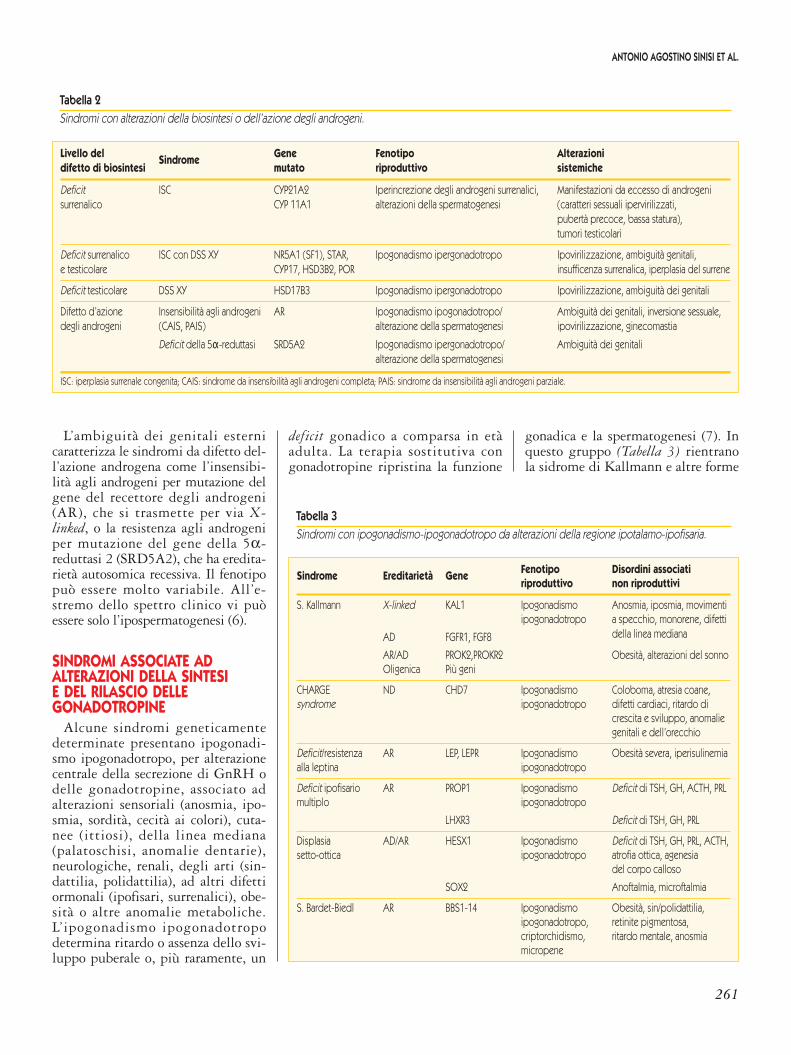
Livello del Sindrome Gene Fenotipo Alterazionidifetto di biosintesi mutato riproduttivo sistemiche
Deficit ISC CYP21A2 Iperincrezione degli androgeni surrenalici, Manifestazioni da eccesso di androgenisurrenalico CYP 11A1 alterazioni della spermatogenesi (caratteri sessuali ipervirilizzati,
pubertà precoce, bassa statura),tumori testicolari
Deficit surrenalico ISC con DSS XY NR5A1 (SF1), STAR, Ipogonadismo ipergonadotropo Ipovirilizzazione, ambiguità genitali,e testicolare CYP17, HSD3B2, POR insufficenza surrenalica, iperplasia del surrene
Deficit testicolare DSS XY HSD17B3 Ipogonadismo ipergonadotropo Ipovirilizzazione, ambiguità dei genitali
Difetto d’azione Insensibilità agli androgeni AR Ipogonadismo ipogonadotropo/ Ambiguità dei genitali, inversione sessuale,degli androgeni (CAIS, PAIS) alterazione della spermatogenesi ipovirilizzazione, ginecomastia
Deficit della 5α-reduttasi SRD5A2 Ipogonadismo ipergonadotropo/ Ambiguità dei genitalialterazione della spermatogenesi
ISC: iperplasia surrenale congenita; CAIS: sindrome da insensibilità agli androgeni completa; PAIS: sindrome da insensibilità agli androgeni parziale.
Tabella 2Sindromi con alterazioni della biosintesi o dell’azione degli androgeni.
Sindrome Ereditarietà Gene Fenotipo Disordini associatiriproduttivo non riproduttivi
S. Kallmann X-linked KAL1 Ipogonadismo Anosmia, iposmia, movimentiipogonadotropo a specchio, monorene, difetti
AD FGFR1, FGF8 della linea mediana
AR/AD PROK2,PROKR2 Obesità, alterazioni del sonnoOligenica Più geni
CHARGE ND CHD7 Ipogonadismo Coloboma, atresia coane,syndrome ipogonadotropo difetti cardiaci, ritardo di
crescita e sviluppo, anomaliegenitali e dell’orecchio
Deficit/resistenza AR LEP, LEPR Ipogonadismo Obesità severa, iperisulinemiaalla leptina ipogonadotropo
Deficit ipofisario AR PROP1 Ipogonadismo Deficit di TSH, GH, ACTH, PRLmultiplo ipogonadotropo
LHXR3 Deficit di TSH, GH, PRL
Displasia AD/AR HESX1 Ipogonadismo Deficit di TSH, GH, PRL, ACTH,setto-ottica ipogonadotropo atrofia ottica, agenesia
del corpo calloso
SOX2 Anoftalmia, microftalmia
S. Bardet-Biedl AR BBS1-14 Ipogonadismo Obesità, sin/polidattilia,ipogonadotropo, retinite pigmentosa,criptorchidismo, ritardo mentale, anosmiamicropene
Tabella 3Sindromi con ipogonadismo-ipogonadotropo da alterazioni della regione ipotalamo-ipofisaria.

Ipogonadismo maschile sindromico
262
di ipogonadismo sindromico, tra cuiricordiamo la sindrome di Bardet-Biedl. Quest’ultima è una sindromecomplessa con alterazioni della vista(retinite pigmentosa) e riproduttive(criptorchidismo, micropene, ipogo-nadismo ipogonadotropo) associate aobesità, ritardo mentale e anomalieneurologiche, sensoriali, nefro-uri-narie, cardiovascolari e ossee, conuna ereditarietà di tipo autosomicorecessiva per mutazione della fami-glia di geni BSS1-14.
Altre sindromi con importantialterazioni neurologiche, sensoriali eintellettive, presentano un ipogona-dismo a patogenesi centrale e perife-rica (Tabella 4). Nella sindrome diPrader-Willi (PWS) si ha obesità,ritardo mentale e disturbi comporta-mentali, microdattilia, strabismo,criptorchidismo, microrchidia, ritar-do o mancata pubertà nei ragazzi. LaPWS è dovuta ad anomalie (delezio-ni, alterazioni dell’imprinting, diso-mia uniparentale) di una regione delcromosoma 15 (15q11-13). Le altera-zioni riproduttive sono dovute aipogonadismo ipogonadotropo inalcuni casi, ma più frequentementeè stato dimostrato un danno tubula-re primitivo che si manifesta dopo lapubertà con l’aumento dell’FSH econ azoo-/oligozoospermia (8).
Un’altra sindrome complessa è ladistrofia miotonica tipo 2, in cui la
disfunzione muscolare (miotonia nel90% dei casi, debolezza e dolorimuscolari nel 10%) si associa a cata-ratta subcapsulare, anomalie di con-duzione cardiaca, diabete tipo 2 eatrofia dei testicoli con aumentodelle gonadotropine .
L’associazione dell’ipogonadismocon l’atassia cerebellare è molto rara.La sindrome compare nell’infanziacon disturbi cortico-spinali progres-sivi, demenza, difetti di crescita sta-turale e ipogonadismo ipogonado-tropo, ma sono descritti casi conipogonadismo ipergonadotropo.
La sindrome di Wolfram è un’altrararissima sindrome in cui l’atrofiaottica e il diabete mellito tipo 1 siassociano ad alterazioni neurodege-nerative e atrofia gonadica. È unamalattia autosomica recessiva dovu-ta a mutazione del gene WFS1.
SINDROMI CON ALTERAZIONIRIPRODUTTIVE POST-TESTICOLARIURO-GENITALI O CON ANOMALIEBIOCHIMICHE E MOLECOLARI DEIGAMETI
Il criptorchidismo è presente indiverse sindromi per alterazionedella discesa della gonade durante lavita embrio-fetale con differentimeccanismi. Si può trovare un elencodelle sindromi con criptorchidismoin pubblicazioni dedicate (9, 10).
L’ostruzione delle vie escretorieseminali può essere presente in alcu-ne sindromi con alterazioni nefro-uri-narie. Le mutazioni del gene CFTR,responsabili dell’assenza bilaterale deidotti deferenti (CBAVD), possonodare in rari casi una sindrome conCBAVD e anomalie renali (assenzadel rene, rene pelvico). Nella malattiapolicistica renale autosomico-domi-nante (ADPKD), caratterizzata dallapresenza di cisti nei reni e in altriorgani (fegato, pancreas, aracnoide),aneurismi intracranici e anomalievalvolari cardiache, vi è azoospermiaper ostruzione cistica dell’ epididimo.
Altre due sindromi con alterazionirenali sono dovute a mutazione delgene WT1 (una dominante, sindro-me di Denys-Drash, e l’altra recessi-va, sindrome di Frasier) e presentanotumore di Wilms, nefropatia, insuf-ficienza renale, ambiguità sessuale egonadoblastoma .
In alcune sindromi l’infertilità èdovuta ad alterazioni funzionalidegli spermatozoi per difetti biochi-mici geneticamente determinati.Nella discinesia ciliare primitiva(PCD), con ereditarietà autosomicarecessiva, si ha situs inversus e altera-zioni del complesso dineinico o del-l’apparato centrale delle struttureciliari, con malattia cronica oto-sino-polmonare e immobilità degli sper-matozoi.
Sindrome Ereditarietà Gene Fenotipo
Ipoplasia surrenale XR AHC, DAX1, NR0B1 Ipogonadismo ipogonadotropo, deficit testicolare,congenita (AHC) insufficienza surrenale, ambiguità genitali
S. Prader-Willi Anomalie della regione critica 15q11-13 Obesità, iperfagia, ritardo mentale, microdattilia,(delezione, difetto di imprinting, strabismo, criptorchidismo, micropenedisomia uniparentale materna)
Distrofia miotonica AD CNBP (ZNF9) Miotonia, debolezza muscolare, cataratta subcapsulare,tipo 2 cardiopatia, alterazioni metaboliche, atrofia testicolare e
ipogonadismo ipergonadotropo
Atassia cerebellare AR Atassia, demenza, sordità, ipogonadismo ipogonadotropoe ipogonadismo o ipergonadotropo, difetti di crescita
S. Wolfram AR WFS1 Atrofia ottica progressiva, diabete mellito tipo 1,(DIDMOAD) alterazioni neurodegenerative ipotalamiche e cerebellari,
sordità, insufficienza renale e atrofia gonadica
Tabella 4Sindromi con ipogonadismo da difetto misto centrale e periferico.

Una sindrome caratterizzata dasordità prelinguale e da alterazionimorfo-funzionali degli spermatozoiè la sindrome sordità-infertilità(deafness-infertility syndrome, DIS),dovuta a delezione di geni continuia livello di 15q15.3 che coinvolge igeni CATSPER2, che causa anoma-lie spermatiche, e STRC, che causala sordità (11).
CONCLUSIONIIn numerose sindromi a eziologia
genetica vi può essere un disordineriproduttivo con ipogonadismo einfertilità nel maschio (1, 10).Queste sindromi vengono diagnosti-cate in età pediatrica per le malfor-mazioni somatiche e sistemiche nonriproduttive (cardiovascolari, nefrou-rinarie, ossee, neurologiche, sensoria-li, metaboliche). L’interessamentoriproduttivo può essere clinicamentemanifesto nell’infanzia come ambi-guità dei genitali, micropene o crip-torchidismo, più tardi come ritardoo maturazione incompleta dellapubertà e in età adulta come deficitdi testosterone, alterazioni delnumero e della funzione degli sper-matozoi. In casi con ipogonadismo oinfertilità sindromici bisogna ricer-care l’eventuale alterazione geneticaresponsabile, non solo per la defini-zione eziologica, ma anche per laprevenzione della trasmissione dellapatologia. La clinica, il tipo di eredi-tarietà e la valutazione dell’assettoendocrino devono indirizzare lo stu-dio genetico-molecolare. Sebbene l’e-lenco delle cause genetiche delle
alterazioni della spermatogenesi siacostantemente in aumento, il poten-ziale coinvolgimento di altri geni eloci rimane sconosciuto. I disordiniriproduttivi sindromici possono aiu-tare a identificare il ruolo di nuovigeni nella riproduzione.
La terapia dell’ipogonadismo sin-dromico si basa sulla somministra-zione sostitutiva di preparati ditestosterone per favorire lo sviluppopuberale ed eliminare i sintomi dacarenza androgenica. Nelle formesecondarie può essere stimolata laspermatogenesi con le gonadotropinequando sia richiesta la fertilità. Ilprelievo dei gameti dai testicoli puòessere indicato in alcuni casi conostruzione o ipospermatogenesirefrattaria alle gonadotropine nel-l’ambito di coppie che vogliono sot-toporsi a tecniche di fecondazioneassistita. Nei soggetti in cui il difet-to testicolare compare o si aggravacon la pubertà (sindrome diKlinefelter, nanismo MUL, PWSecc.) si discute se un prelievo di tes-suto gonadico in età adolescenzialepossa permettere di ottenere celluledella linea spermatogenetica da con-gelare per l’uso nella fertilizzazionein vitro in età adulta. Tuttavia, ipochi dati attualmente disponibili(in particolare in adolescenti con sin-drome di Klinefelter) non sembranofavorevoli a un recupero di celluledella spermatogenesi (12).
BIBLIOGRAFIA1. Krausz C 2010 Male infertility: pathogenesis
and clinical diagnosis. Best Pract Clin Endo-crinol Metab 25:271-285.
2. Vorona E, Zitzmann M, Gromoll J,Schüring AN, Nieschlag E 2007 Clinical,endocrinological, and epigenetic features of the46,XX male syndrome, compared with47,XXY Klinefelter patients. J Clin EndocrinolMetab 92: 3458-3465.
3. Ankarberg-Lindgren C, Westphal O, Dahl-gren J 2011. Testicular size development andreproductive hormones in boys and adult maleswith Noonan syndrome: a longitudinal study.Eur J Endocrinol 165:137-144.
4. Karlberg S, Toppari J, Karlberg N et al 2011testicular failure and male infertility in themonogenic mulibrey nanism disorder. J ClinEndocrinol Metab 96:3399-3407.
5. Reisch N, Flade L, Scherr M et al 2009 Highprevalence of reduced fecundity in men withcongenital adrenal hyperplasia. J ClinEndocrinol Metab 94:16665-1670
6. Gottlieb B, Beitel LK, Trifiro MA 2011Androgen insensitivity syndrome.GeneReviews™.
7. Sinisi AA, Maione L, Bellastella G, Asci R,Bellastella A 2011 Diagnosi e terapia dell’i-pogonadismo nella sindrome di Kallmann.L’Endocrinologo 12:8-19.
8. Siemensma EPC, de Lind van WijngaardenRFA, Otten BJ, de Jong FH, Hokken-Koelega ACS 2012 Testicular failure in boyswith Prader-Willi syndrome: longitudinalstudy of reproductive hormones. J ClinEndocrinol Metab 97:E452-E459.
9. Foresta C, Zuccarello D, Grolla A, Ferlin A2008 Role of hormones, genes, and environ-ment in human cryptorchidism. Endocrine Rev29:560-580.
10. Lyons Jones K 1997 Smith’s recognizable pat-terns of human malformation. Philadelphia-Tokio: WB Saunders Co.
11. Hidebrand MS, Avenarius MR, Smith RJH2009 CATSPER-related male infertility.GeneReviews http://www.geneclinics.org/profiles/all.html
12. Gies I, De Schepper J, Van Saen D,Anckaert E, Goossens E, Tournaye H 2012Failure of a combined clinical and hormonalbased strategy and retrieve spermatogonial stemcells in 47,XXY boys by single testicular biop-sy. Hum Reprod 27:998-1004.
263
ANTONIO AGOSTINO SINISI ET AL.


















