
Epilessia - ARNo Neurologia | Associazione per la Ricerca...
43
EPILESSIA
Transcript of Epilessia - ARNo Neurologia | Associazione per la Ricerca...

EPILESSIA

EPILESSIA
CRISI EPILETTICAManifestazione clinica parossistica dovuta ad una scarica anomala, ipersincrona di una popolazione più o meno estesa di neuroni corticali.
Epileptic Seizures and Epilepsy: Definitions Proposed by the International League Against Epilepsy (ILAE) and the International Bureau for Epilepsy !“Disorder of the brain characterized by an enduring predisposition to generate epilettic seizures and by the neurobiologic, cognitive, psycological, and social consequences of this condition. The definition of epilepsy requires the accurrence of at least on epilettic seizure.” Fisher et al. 2005
Malattia cronica caratterizzata dalla ricorrenza di crisi epilettiche non provocate. Un cluster di crisi manifestatesi nelle 24 h equivale ad una singola crisi.

CRISI EPILETTICA NON PROVOCATA
Crisi che occorre in assenza di fattori scatenanti, sono escluse crisi da danno acuto del SNC o da insulto metabolico sistemico (crisi sintomatica acuta), sono invece comprese le crisi da abnorme sensibilità a stimoli esterni (es. crisi fotoconvulsive).
CRISI EPILETTICA SINTOMATICA ACUTA
Crisi che si manifesta in stretta associazione temporale con danno acuto sistemico, metabolico o tossico del SNC e che si presume sia la conseguenza diretta del danno.
CRISI EPILETTICA SINTOMATICA REMOTA
Crisi sintomatica occorsa in soggetti con danno non recente del SNC. Definizione limitata a fattori antecedenti per i quali sia chiaramente documentato un potenziale epilettogeno.

Epilessia !50 milioni di individui nel mondo soffrono di epilessia !Di questi il 75% si trovano in paesi in via di sviluppo (9 su 10 non ricevono un trattamento adeguato) !Incidenza 40-70/100 000 (paesi industrializzati) Prevalenza 4-10/100 000 (paesi industrializzati) !Molti studi mostrano che l'epilessia è più frequente nel sesso maschile che nel sesso femminile !Incidenza uomo: 50,7/100 000 Incidenza donna: 46,2/100 000 !! !!
EPIDEMIOLOGIAPrima crisi !Incidenza di una prima crisi non provocata 50-70/100 000 ! - paesi industrializzati 40-70/100 000 - paesi in via di sviluppo 100-190/100 000
UCL Istitute of Neurology Queen Square, London

Epilessia – distribuzione per età
•La curva specifica di incidenza per età dell'epilessia ha una distribuzione bimodale. !•Elevata nel primo anno di vita con relativa riduzione durante l'adolescenza. !•Il più basso tasso di incidenza è fra I 20 e I 40 anni. !•L'incidenza diventa particolarmente elevata dopo gli 80 anni.
UCL Istitute of Neurology Queen Square, London
EPIDEMIOLOGIA

Data from Mortality and Burden of Disease estimates for WHO member states in 2002
Age-standardised disability-adjusted life year (DALY) rates from Epilepsy by country (per 100,000 inhabitants)
EPIDEMIOLOGIA

FISIOPATOLOGIA
!!
● un'eccessiva e abnorme attività (scarica epilettica) sia di singoli che di grossi gruppi di neuroni (focalaio epilettico)
!● uno sbilanciamento fra neurotrasmettitori eccitatori ed inibitori
!● alterazione dei sistemi corticali e sottocorticali
Alla base della crisi epilettica vi è:

Variazione temporanea e parossistica dell'attività dell'EEG, focale o diffusa. !La genesi di una scarica epilettica implica l'esistenza di un disturbo costituzionale o acquisito
dell'eccitabilità neuronale.
!Neuroni epilettici: !Ipereccitabilità: tendenza di un neurone a generare scariche ripetute in risposta stimoli che,
in condizioni normali, dovrebbero indurre un solo potenziale d'azione.
!Ipersincronia: capacità di un gruppo di neuroni di generare una serie di potenziali d'azione in
maniera sincrona.
SCARICA EPILETTICA I

NEURONE NORMALE vs EPILETTICO
Un singolo neurone epilettico non è in grado di reclutare i neuroni normali circostanti. Solo una scarica epilettica parossistica, di un aggregato neuronale corticale di sufficiente dimensione (massa critica), riesce a produrre manifestazioni focali intrinseche. !!Quando la scarica di burst coinvolge non una singola cellula ma un aggregato neuronale si può parlare di situazione epilettiforme.
Un'attività di scarica di tipo epilettico è legata alla modificazione transitoria delle proprietà di membrana ed è espressa dal fenomeno di brusca depolarizzazione di membrana detto paroxymal Depolarization shift (PDS). !!Paroxymal Depolariation Shift o PDS si esprime come una repentina scarica di potenziali d'azione ad alta frequenta, multipli e ripetitivi (burst) di notevole ampiezza (20-40 microV) e durata fino a 250 msec, che si interrompe quando le difese inibitorie entrano in azione.
SCARICA EPILETTICA II

FOCOLAIO EPILETTICONel contesto del focolaio epilettogeno esistono: !Neuroni fortemente epilettici - dotati di scarica ricorrente stereotipata o evocata da stimoli afferenti – burst di 8-15 spikes ad elevata frequenza molto spesso intervallati da intervalli lunghi. !Neuroni parzialmente epilettici - suscettibili di una certa modulazione da parte di stimoli afferenti, sonno o veglia. !!Neuroni normali (suscettibili di rapido reclutamento in un pattern di scarica patologica ipersiscrona ad alta frequenza in particolari circostanze favorenti). !Solo nel caso di un reclutamento di un sufficiente numero di neuroni è possibile registrare Complesso punta onda.

SBILANCIAMENTO DEI SISTEMI ECCITATORI ed INIBITORI
Inibizione !GABA Eccitazione
!Glutammato Aspartato
Modified from White, 2001

Due gruppi di recettori del glutammato !Inotropi (iGlu) – trasmissione sinaptica rapida:
R AMPA
R del Kainato
R NMDA (N-metil-D-aspartato)
!Metabotropi – trasmissione sinaptica lenta:
mGluR (mGlu1 - mGlu8)
Principale neurotrasmettitore eccitatorio del SNC
GLUTAMMATO

!Determina iperpolarizzazione della membrana neuronale
I recettori GABA A sono il Recettori del GABA: !Inotropi:
GABA A – post-sinaptici; selettivemente
permiabile al Cl-
GABA C - esclusivo della retina !Metabotropo:
GABA B – pre-sinaptici; azione inibitoria
più lenta; riduzione dell'afflusso
di calcio e aumento della corrente del K+
bersaglio di numerose sostanze neuroattive che si legano a siti recettoriali specifici (bdz, barbiturici, alcool, neurosteroidi ..)
GABAPrincipale neurotrasmettitore inibitorio del SNC

Ioni Na+ Ca++Responsabili della corrente di depolarizzazione
!!
Generazione del potenziale di azione
Ioni K+ Cl-Responsabili della ripolarizzazione
!!
Inibizione del potenziale di azione
MECCANISMI IONICI

In base all'eziopatogenesi vengono distinte: !Crisi idiopatiche: Presunte genetiche, che compaiono indipendentemente da ogni lesione cerebrale. Il cui principale fattore eziologico è rappresentato da una predisposizione genetica reale o presunta. !Crisi sintomatiche: Da una lesione strutturale diffusa o focale, fissa o evolutiva, documentabile a carico del SNC. !Crisi criptogenetiche: Si ritiene siano sintomatiche di una causa occulta la quale sfugge ai nostri mezzi di investigazione. Epilessia senza lesioni evidenti ma che non corrisponde ai criteri di epilessia idiopatica. Nella proposta di revisione della terminologia ILAE 2010 vengono indicati nuovi termini e concetti: !
CLASSIFICAZIONE EZIOLOGICA
Idiopatiche -> Genetica: un difetto genetico è causa diretta dell'epilessia e le crisi sono sintomo principale del disturbo. !Sintomatiche -> Strutturali-metaboliche: causate da una disordine metabolico o strutturale del cervello. !Criptogenetiche -> eziologia sconosciuta: la causa è sconosciuta e potrebbe essere genetica, strutturale o metabolica.

EZIOLOGIA (1)• Le eziologie delle epilessie sono molto eterogenee e risultano dalla
combinazione di fattori genetici e di fattori acquisiti. In differenti circostanze uno di questi può predominare.
!•Nelle epilessie con più forte determinismo genetico i fattori esogeni favoriscono l'espressione della patologia. !•Analogamente i fattori genetici determinano molto probabilmente il potenziale epilettogeno di lesioni strutturali del SNC.
FATTORI GENETICI: !
● Anomalie cromosomiche (es. Sindrome di Down, Sindrome dell' X-fragile, Sindrome di Angelman ecc..)
!● Mutazioni di singoli geni (epilessie monogeniche) possono essere autosomiche
dominanti, recessive, X-linked o mitocondriali (mDNA). ● Poligenetiche nella maggioranza delle sindromi epilettiche idiopatiche.

EZIOLOGIA (2)!
FATTORI ACQUISITI: !● Fattori perinatali !● Anomalie dello sviluppo corticale !● Lesioni cicatriziali (es. sclerosi dell'ippocampo) !● Malattie infettive: Encefaliti virali, meningoencefaliti batteriche, ascessi cerebrali, neurocistercosi, HIV
(neurotropismo virale o infezione opportunistica), meningite tubercolare, sifilide. !● Tumori cerebrali responsabili del 10-15% delle crisi nell'adulto. !● Traumi cranici
- crisi precoci (entro una settimana dal trauma) - crisi tardive (epilessia post-traumatica o EPT propriamente detta) !
● Malattie cerebro-vascolari rappresentano la causa principale di epilessia nell'anziano. !● Malformazioni cerebro-vascolari (es. MAV, cavernomi) !● Vasculiti cerebrali LES, vasculiti complicanti la tossiemia gravidica. !● Fattori tossici (Alcool, cocaina, anfetamine, fenciclidina, piombo, manganese, metanolo, organofosfati..) !● Fattori farmacologici per impregnazione cronica (es. antipsicotici), sovradosaggio (es. carbonato di litio) o
astinenza (es. barbiturici, bdz). !● Fattori metabolici (modificazioni acute dell'equilibrio elettrolitico, ipoglicemia ed iperglicemia con iperosmolarità,
encefalopatie uremiche).

CLASSIFICAZIONE DELLE CRISI EPILETTICHE – ILAE 1981
1. CRISI PARZIALI (FOCALI, LOCALIZZATE) !!A. crisi parziali semplici (senza disturbo della coscienza)
● Con segni motori:motorie focali con e senza marcia, versive, posturali, fonatorie
● Con sintomi somatosensoriali o sintomi sensoriali particolari:somatosensitive, visive, uditive, olfattive, gustative, vertiginose, allucinatorie semplici
● Con manifestazioni autonomiche, inclusa “aura epigastrica”
● Con manifestazioni psichiche (dist.funzioni corticali superiori): disfasiche, dismnesiche, con dist.cognitivi ,affettivi, con illusioni, anche complesse !
B. crisi parziali complesse ● Inizio parziale semplice seguito da disturbo di
coscienza - con caratteristiche parziali semplici seguite da
sola compromissione della coscienza - con automatismi
● Con iniziale compromissione della coscienza - solo con compromissione della coscienza - con automatismi !
C. crisi parziali che evolvono in crisi generalizzate ● Crisi parziali semplici ● Crisi parziali complesse ● Crisi parziali semplici – parziali complesse – crisi
secondariamente generalizzate
2. CRISI GENERALIZZATE !!A. crisi a tipo assenza
● Solo con compromissione della coscienza, componenti cloniche modeste, componenti atoniche, componenti toniche, automatismi, componenti autonomiche.
● Crisi tipo assenza atipica con modificazione del tono più pronunciata rispetto ad 1., e con esordio e\o termine progressivi. !
B. crisi miocloniche C. crisi cloniche !D. crisi toniche !E. crisi tonico-cloniche !F. crisi atoniche
3. CRISI NON CLASSIFICABILI

CLASSIFICAZIONE DELLE CRISI EPILETTICHE
1. Berg AT et al. Revised terminology and compets for organization of seizures and epilepsies: report of the ILAE Commission on Classification and Terminology, 2005-2009. Epilessia 2010; 51: 676-685. 2. Berg AT, Cross JH. Lancet 2010; 9: 459-61. 3. Blume WT et al. Glossary of descriptive terminology for ictal semiology: Report of the ILA ask force on classification and terminology. Epilessia 2001: 42; 1212-1218.
ILAE proposta di revisione della terminologia per l'organizzazione delle crisi e delle epilessia 2010

● Tonico-cloniche !● Assenza
- Tipiche - Atipiche !
● Cloniche !● Toniche !● Atoniche !● Miocloniche
- Micloniche - Miocloniche-atoniche - Micloniche-toniche
La scarica parossistica è fin dall'inizio estesa ai due emisferi e sembra coinvolgere simultaneamente, in questo modo, l'insieme della corteccia cerebrale
CRISI GENERALIZZATE

Crisi tonico-cloniche (grande male)Sono le crisi generalizzate più frequenti. Si manifestano in tre fasi: tonica, clonica, risolutiva. !Fase tonica
● Da 10 a 20 secondi. ● L'esordio è spesso caratterizzato da un grido. ● La perdita di coscienza è immediata. ● Una contrazione tonica sostenuta interessa l'insieme della muscolatura scheletrica. ● Sono associate importanti turbe vegetative (tachicardia, aumento della pressione
arteriosa, midriasi, arrossamento del volto, ipersecrezione salivare e bronchiale). ● Talvolta si osserva una morsicatura laterale della lingua. !
Fase clonica ● Circa 30 secondi. ● Il rilasciamento intermittente della contrazione muscolare tonica determina scosse
bilaterali, sincrone, intense, che si distanziano progressivamente fino ad interrompersi bruscamente.
● Lo stato apnoico che si produce all'inizio della crisi determina un'intensa cianosi del volto.
!Fase post-critica (o risolutiva)
● Da alcuni minuti ad alcune decine di minuti. ● Il soggetto si presenta ipotonico, immobile, con profonda obnubilazione della coscienza e un rilasciamento muscolare completo. ● Possono esserci enuresi ed encopresi ● La respirazione riprende ampia e rumorosa, disturbata dall'ipersecrezione bronchiale e salivare. !
Quando il soggetto non si addormenta, il livello di coscienza migliora progressivamente, l'obnubilamento lascia il posto ad una confusione mentale di intensità e durata variabili, talvolta accompagnata da automatismi. !Alla fine della crisi o al risveglio il paziente lamenterà spesso cefalea, spossatezza e dolori muscolari (talvota correlati ad un'innalzamento degli enzimi muscolari).
Clinica Symposia Ciba 1994

AssenzeLe assenze sono crisi di breve durata, in cui la manifestazione principale è un'alterazione (attenuazione o sospensione) della coscienza.
Assenze tipiche (piccolo male) ● Durata di 5 – 30 secondi. ● Esordio e fine bruschi. ● Alterazione isolata della coscienza; un soggetto si immobilizza, con occhi fissi nel vuoto,
interrompendo l'attività in corso. ● Al termine della crisi il soggetto riprende la propria attività e non conserva alcun ricordo
dell'episodio. ● Si accompagnano a una scarica bilaterale, sincrona e simmetrica di punte-onde la cui frequenza è
superiore o uguale a 3 Hz, questa scarica sopravviene su un attività di fondo normale. ● Osservabili in epilessie idiopatiche dell'infanzia. !
Assenze atipiche ● Durata generalmente maggiore delle assenze tipiche. ● Esordio e fine più progressivi. ● L'alterazione della coscienza spesso è meno marcata. ● Sono spesso presenti elementi tonici e/o mioclonici più pronunciati e meno simmetrici. E' anche
possibile osservare una caduta lenta del corpo, progressiva, abitualmente non traumatica. ● Si accompagnano a scariche di punte-onde bilaterali regolari, talvolta sincrone, di frequenza inferiore
ai 3 Hz che possono essere associate ad altri aspetti critici. Queste scariche compaiono su un attività di fondo che è spesso anormale.
● Osservabili nelle epilessie sintomatiche dell'infanzia !!

Crisi clonicheScosse cloniche bilaterali, asimmetirce, progressivamente più lente e durata variabile, alle quali concomita un'alterazione della coscienza e obnubilamento post-critico. Elettivamente nella prima e nella seconda infanzia, talvolta nell'ambito di una convulsione febbrile.
Crisi tonicheSi caratterizzano per una contrazione muscolare sostenuta, non vibratoria, protratta per almeno alcuni secondi, si associa ad alterazione della coscienza, a un'apnea e ad altre manifestazioni vegetative. Spesso in serie.
Crisi atonicheSi caratterizzano per una risoluzione del tono posturale, ne consegue una caduta violenta e traumatizzante. Talvolta si limita ad una semplice caduta del capo in avanti.
Crisi miclonicheSul piano neurofisiologico si caratterizzano per una contrazione simultanea di muscoli agonisti e antagonisti che determinano una scossa improvvisa e breve. Queste scosse interferiscono con la funzione motoria, il coinvolgimento degli arti inferiori può determinare cadute. Le crisi miocloniche generalizzate si presentano come scosse bilaterali e simmetriche che compaiono in assenza di alterazioni percettibili della coscienza.

CRISI FOCALI
Le crisi verrebbero distinte in base alle specifiche caratteristiche semiologiche; presenza di una o più caratteristiche quali: - aura - motorie - autonomiche - coscienza/responsività: alterata (discognitive ex parziali complesse) o mantenuta (ex crisi parziali semplici)
Nella proposta di revisione della terminologia delle crisi epilettiche ILAE 2010 viene sconsigliato l'utilizzo dei termini: - crisi parziali semplici - integrità della coscienza - crisi parziali complesse - presenza di un disturbo della coscienza iniziale o secondario
Possono evolvere in crisi convulsive bilaterali o secondariamente genralizzate.
La semeiologia clinica delle crisi parziali dipende strettamente e direttamente dalle caratteristiche anatomo-funzionali del circuito epilettogeno, e cioè dall’area di partenza (importanza del sintomo iniziale) e dalle strutture via via reclutate
Che originano con circuiti limitati ad un singolo emisfero.
Benigne Catastrofiche !Parziali complesse Parziali semplici !Secondariamente generalizzate
Autolimitanti: tendenti a risolversi spontaneamente col tempo Farmacoresponsive: alta probabilità di controllo con farmaci
Crisi focali: semiologia delle crisi descritta sulla base di specifiche caratteristiche soggettive, motorie, autonomiche e discognitive
Evoluzione in crisi bilaterali convulsive: per es. tonico, cloniche, tonico-cloniche

Crisi focali con sintomi motori o somatomotorieCrisi somatomotorie con marcia jacksoniana (sono le sole crisi attribuibili ad un origine corticale precisa) La scarica epilettica unidirezionale, progredisce sulla corteccia rolandica motoria restandovi circoscritta. Clinicamente: contrazione tonica seguita da scosse cloniche che esordisce in una porzione limitata di un arto (pollice, alluce) o del volto e si estende per contiguità.
Crisi somatomotorie senza marcia jacksoniana Combinazione variabile di manifestazioni cloniche e/o toniche di una parte più o meno estesa di un emisoma. La scarica disorganizza frequentemente sia la corteccia motoria primaria che le regioni premotorie.
Crisi con carattere tonico-posturale, talvolta distonico – orienta per un'origine più frontale delle scariche, sebbene l'assunzione di una postura anomala di un arto possa essere associata anche ad una scarica critica della corteccia parietale. !Crisi versive – deviazione della testa e/o degli occhi (lenta o rapida, saccadica o tonica, coniugata o meno). Possono associarsi a rotazione del tronco (origine premotoria dorsolaterale), ad elevazione ed abduzione dell'arto superiore omolaterale, arresto del linguaggio o a vocalizzazione (area supplementare motoria) !Crisi fonatorie – impossibilità ad emettere parole o vocalizzazioni.
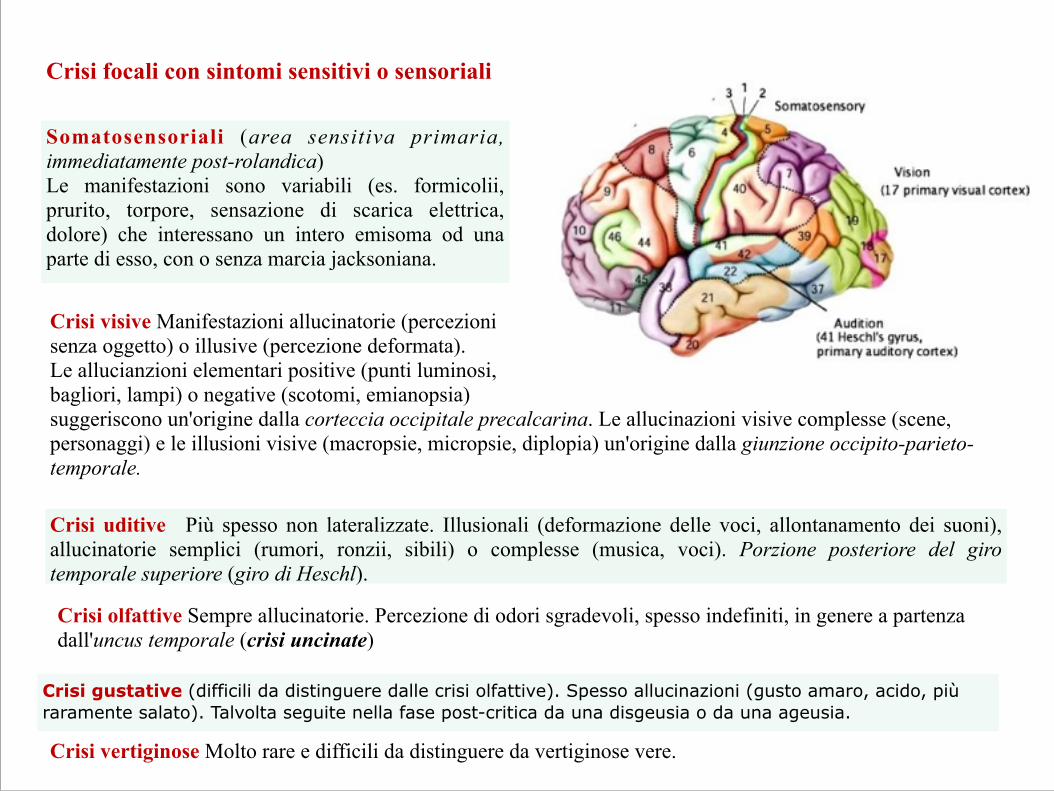
Crisi focali con sintomi sensitivi o sensoriali
Somatosensoriali (area sensitiva primaria, immediatamente post-rolandica) Le manifestazioni sono variabili (es. formicolii, prurito, torpore, sensazione di scarica elettrica, dolore) che interessano un intero emisoma od una parte di esso, con o senza marcia jacksoniana.
Crisi visive Manifestazioni allucinatorie (percezioni senza oggetto) o illusive (percezione deformata). Le allucianzioni elementari positive (punti luminosi, bagliori, lampi) o negative (scotomi, emianopsia) suggeriscono un'origine dalla corteccia occipitale precalcarina. Le allucinazioni visive complesse (scene, personaggi) e le illusioni visive (macropsie, micropsie, diplopia) un'origine dalla giunzione occipito-parieto-temporale.
Crisi uditive Più spesso non lateralizzate. Illusionali (deformazione delle voci, allontanamento dei suoni), allucinatorie semplici (rumori, ronzii, sibili) o complesse (musica, voci). Porzione posteriore del giro temporale superiore (giro di Heschl).
Crisi olfattive Sempre allucinatorie. Percezione di odori sgradevoli, spesso indefiniti, in genere a partenza dall'uncus temporale (crisi uncinate)
Crisi gustative (difficili da distinguere dalle crisi olfattive). Spesso allucinazioni (gusto amaro, acido, più raramente salato). Talvolta seguite nella fase post-critica da una disgeusia o da una ageusia.
Crisi vertiginose Molto rare e difficili da distinguere da vertiginose vere.

Sfera digestiva Sensazioni anali, addominali digestive, esofagee o faringee, di carattere variabile, localizzate o migranti (sensazione di pesantezza, costrizione, dolore, calore, nausea ecc..); più frequenti per le crisi temporali. Eruttazioni, vomito, atto di sputare o ipersecrezione salivare (regione opercolo insulare). !Tratto urogenitale Bisogno o atto di urinare (cortecia orbiataria), più raramente sensazione genitale, rarissimi erezione e orgasmo. !Sistema cardiovascolare Palpitazioni, arrossamento, pallore, tachicardia o bradicardia. !Sistema respiratorio Sensazione di soffocamento, tosse, difficoltà respiratorie, apnea, bradipnea, polipnea. !Termoregolazione Brividi, pelle d'oca, ondate di calore, sudorazione. !Muscolatura oculare intrinseca midriasi, molto più rara miosi.
Crisi focali con manifestazioni vegetative

Si caratterizzano per un'alterazione elettiva delle funzione corticali superiori. !Oltre ai fenomeni illusionali, allucinatori e afasici, queste manifestazioni “esperenziali” combinano tipicamente elementi percettivi, mnesici e affettivi. !Dreaming state (o stato sognante) con questo termine vengono raggruppati diversi fenomeni legati alla disorganizzazione delle strutture temporali mesiali e laterali aventi in comune una percezione alterata della realtà ambientale. Ad esempio impressione di estraneità, irrealtà o vissuto del presente con modalità oniriche: - allucinazioni “esperenziali” con ricordo forzato e inadatto di avvenimenti passati, - diplopia mentale il pz si sente nel presente mentre vive simultaneamente una scena del passato. - illusione di familiarità (déjà-vu, déjà-vécu) o illusione di estraneità (jamais-vu, jamais- vécu) si possono spiegare come la percezione differita di informazioni normalmente codificate, con conseguente falsa interpretazione del presente. !Altre manifestazioni come il pensiero forzato (idea parassita che si impone al soggetto), suggerisce una localizzazione frontale. !Disturbo istintivo-affettivi raggruppano diverse sensazioni, più spesso spiacevoli come ansia, paura o vero terrore. In altre occasini irritabilità e aggressività. !Crisi di riso (crisi gelastiche) o di pianto (diacristiche) devono far ricercare un'amartoma ipotalamico.
Crisi focali con sintomi psichici

CRISI PARZIALI COMPLESSE
L'esistenza di un disturbo della coscienza, iniziale o secondario, definisce le crisi parziali complesse. !Si accompagnano spesso ad attività automatiche, che corrispondono a manifestazioni motorie involontarie più o meno elaborate: !
● Automatismi gestuali semplici, unilaterali o bilaterali, diretti verso il paziente o verso l'ambiente circostante. Spesso senza finalità precisa (atto di pedale, dondolamento del braccio, ecc..).
!● Automatismi gestuali complessi realizzano sequenze più elaborate
(abbottonare, sbottonare vestiti, cercare nelle tasche, mettere apposto ecc..). !● Automatismi verbali (onomatopee, esclamazione, parole o frammenti di frasi
ecc..) !

CLASSIFICAZIONE DELLE SINDROMI ELETTROCLINICHE – ILAE 1989
1. GENERALIZZATE !A. Epilessie generalizzate idiopatiche con esordio età-dipendente (in ordine di età)
● Convulsioni neonatali benigne familiari e non familiari
● Epilessia mioclonica benigna dell’infanzia ● Epilessia tipo assenza dell’infanzia ● Epilessia tipo assenza giovanile ● Epilessia mioclonica giovanile ● Epilessia con crisi generalizzate tonico-
cloniche al risveglio ● Altre epilessie generalizzate idiopatiche non
inquadrabili nelle precedenti categorie ● Epilessie con crisi scatenate da modalità
specifiche di attivazione !B. Epilessie generalizzate criptogeniche o sintomatiche (in ordine di età)
● Sindrome di West ● Sindrome di Lennox-Gastaut ● Epilessia con crisi mioclono-astatiche ● Epilessia con assenze miocloniche !
C. Epilessie generalizzate sintomatiche ● Eziologia non-specifica ● Encefalopatia mioclonica precoce ● Encefalopatie epilettiche infantili precoci con
suppression-burst ● Altre epilessie generalizzate sintomatiche non
inquadrabili nelle precedenti categorie ● Sindromi specifiche
2. FOCALI !A. Epilessie localizzate idiopatiche con esordio legato all’età
● Epilessia benigna dell’infanzia con punte centro-temporali ● Epilessia dell’infanzia con parossismi occipitali ● Epilessia primaria da lettura !
B. Epilessie localizzate sintomatiche ● Epilessia parziale continua ● Sindromi caratterizzate da crisi con specifiche modalità di attivazione ● Epilessia del lobo temporale ● Epilessie del lobo frontale ● Epilessie del lobo parietale ● Epilessie del lobo occipitale !
C. Epilessie localizzate criptogeniche
3. EPILESSIE E SINDROMI INDETERMINATE SE FOCALI O GENERALIZZATE !A. Sia con crisi generalizzate che focali
● Crisi neonatali ● Epilessia mioclonica grave dell’infanzia ● Epilessia con punta-onda continua durante il sonno lento (ESES) ● Afasia epilettica acquisita (Sindrome di Landau-Kleffner) !
B. Altre indeterminate generali/ focali
4. SINDROMI SPECIALI !A. Convulsioni febbrili !B. Crisi isolate o stati epilettici isolati !C. Crisi da alterazione acuta metabolica o tossica (alcool, farmaci, eclampsia, iperglicemia non chetosica)
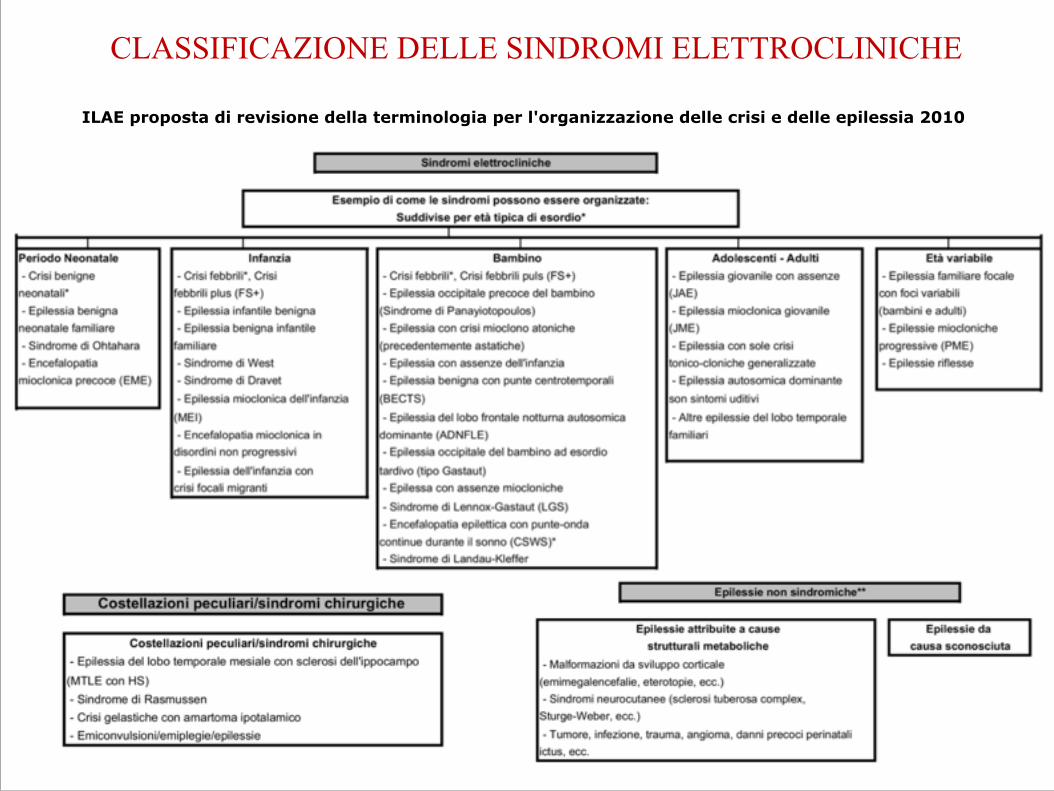
CLASSIFICAZIONE DELLE SINDROMI ELETTROCLINICHE
ILAE proposta di revisione della terminologia per l'organizzazione delle crisi e delle epilessia 2010

Epilessia parziale idiopatica del bambino a parossismi occipitali Comprende due forme cliniche: la Sindrome di Panayiotopoulos o epilessia occipitale a esordio precoce e la Sindrome di Gastaut o epilessia occipitale a esordio tardivo. !Sindrome di Panayiotopoulos o epilessia occipitale ad esordio precoce Età d'esordio compresa tra 3 e 6 anni. Prevalentemente durante il sonno. Colpiscono maschi e femmine alla stessa maniera. ̀Presente un’alta incidenza di familiarità per CF. Evoluzione benigna, tende a risolversi un paio d’anni entro l'adolescenza. !CLINICA: In due terzi dei casi durante il sonno il paziente si sveglia, è vigile ma sofferente, pallido lamenta dolore e nausea, presenta deviazione laterale dello sguardo. Possono essere presenti altri sintomi vegetativi quali il rash cutaneo, cianosi, midriasi o miosi, alterazioni cardio-respiratorie e della termoregolazione, scialorrea e incontinenza. Può subentrare la perdita di coscienza con clonie emifacciali, mioclonie, nistagmo e automatismi. La crisi culmina con il vomito nella maggior parte dei casi. Di solito l’episodio tende a durare a lungo anche più di mezz’ora-un’ora, determinando una sorta di stato di male. Alla fine dell’evento critico il paziente si riprende senza ricordare nulla, dopo qualche ora di sonno. !TRACCIATO EEG: cariche di punte onde lente ad alto voltaggio che possono avere origine in qualsiasi sede, ma hanno una netta prevalenza in regione occipitale. Queste alterazioni si attivano sempre con il sonno. !Sindrome di Gastaut o epilessia occipitale ad esordio tradivo Più rara. Età di esordio fra i 3 e i 15 anni (picco 8 anni). Evoluzione benigna, tende a risolversi pochi anni dopo l'esordio. !CLINICA: Crisi convulsive che si presentano sottoforma di allucinazioni visive e cecità insieme o alternate, della durata di pochi secondi o di qualche minuto. Le allucinazioni visive sono caratteristiche in quanto prevalentemente determinate dalla visione di numerose palline colorate a volte accompagnate da sensazione illusoria di movimento degli occhi o di deviazione da un lato o di dolore. La coscienza non è quasi mai compromessa. La crisi è seguita da cefalea diffusa o localizzata a un emilato, della durata di circa un’ora. (d/d crisi emicranica, epilessia occipitale sintomatica). !TRACCIATO: L’EEG in fase intercritica può essere normale o mostrare le scariche di punte onde lente prevalenti in regione occipitale. L’EEG critico mostra le scariche di attività rapida in regione occipitale. !

Epilessia mioclonica giovanile Esordio fra i 6 e i 25 anni (picco fra i 12 e i 17 anni). !Il quadro clinico è molto caratteristico. Sono presenti scosse miocloniche spontanee, bilaterali, quasi sempre simmetriche, isolate o ripetitive, che coinvolgono gli superiori e la faccia, quando coinvolgono gli arti inferiori possono determinare cadute. Le mioclonie sopravvengono poco dopo il risveglio in piena coscienza e sono tipicamente favorite dalla mancanza di sonno. Nel 90 % dei casi si associano crisi generalizzate tonico-cloniche che esordiscono tipicamente con un crescendo di mioclonie massive bilaterali. Inoltre possono essere presenti assenze brevi molto poco evidenti. !L'epilessia mioclonica giovanile è determinata geneticamente. Più geni contribuiscono verosimilmente a determinare la suscettibilità. (due geni sono verosimilmente coinvolti 6p21.2 e 15q14) !La risposta al trattamento è molto buona ma spesso va protratta per molti anni o per tutta la vita per la comparsa di recidive cliniche nel 90% dei casi. !Epilessia con crisi di Grande Male al risveglio disce durante l'adolescenza. Più frequente nel sesso femminile. Le crisi sopravvengono esclusivamente o in modo predominante poco dopo il risveglio, al mattino, dopo una pisolino o durante il periodo di rilassamento serale. Fattori scatenanti sono la privazione di sonno, assunzione eccessiva di alcol, i risvegli provocati. Un'esacerbazione della frequenza delle crisi si osserva nel periodo catameniale.

SINDROMI SPECIALIConvulsioni febbrili Per definizione è un evento associato a febbre senza segni di infezione intracranica o di altra causa definita, che compare in un lattante o in un bambino di età compresa fra 3 mesi e 5 anni. Le crisi convulsive con febbre che sopraggiungono in bambini che hanno un anamnesi positiva per una crisi epilettica non febbrile sono escluse da questa definizione. Dal 2 al 5% dei bambini sotto i 5 anni presenteranno almeno una crisi febbrile; ma soltanto il 5% di questa popolazione presenterà successivamente un'epilessia propriamente detta. !Convulsioni febbrili “semplici” Hanno prognosi eccellente, compaiono dopo il primo anno di vita. Risultano da una suscettibilità genetica età-dipendente alla febbre. Crisi bilaterali cloniche o tonico-cloniche della durata inferiore ai 15 minuti, che non si ripetono in corso di un episodio febbrile e non presentano segni localizzatori critici o post-critici. Tipicamente dopo 24 h dall'esordio della febbre (nel 95% dei casi di origine virale), durante il picco febbrile o durante il calo termico. !Crisi convulsive febbrili “complicate” Prima del compimento del primo anno di vita in bambini con antecedenti familiari. Convulsioni asimmetriche o unilaterali, cloniche della durata superiore ai 15 minuti, spesso in salve nel corso di un singolo episodio febbrile. Sono seguite da un deficit post-critico di intensità variabile. (Questo crisi rappresenta in effetti un vero e proprio stato di male febbrile). !Il rischio di epilessia successiva è direttamente proporzionale al numero di questi criteri di gravità, se sono presenti almeno tre il rischio è circa il 50%. Il rischio di sviluppare successivamente un'epilessia della porzione mesiale del lobo temporale sarebbe direttamente correlato alla durata delle convulsioni febbrili.

● Stress ● Privazione di sonno e affaticamento ● Disturbo del ciclo sonno/veglia ● Abuso o deprivazione di alcool ● Alterazioni metaboliche ● Fattori tossici e farmaci ● Ciclo mestruale ● Stimolazioni luminose o acustiche o sensoriali in genere
FATTORI PRECIPITANTI UNA CRISI EPILETTICA

DIAGNOSI
SINDROMICA
Anamnesi EEG:
- EEG intercritico in veglia o in sonno - EEG critico - EEG Holter - Video EEG - Registrazione poligrafica
EZIOLOGICA
TC RM, AngioRM
SPECT – PET
Test genetici
Esami ematochimici
Test Neuropsicologici
La diagnosi di epilessia si basa soprattutto sulla descrizione dettagliata delle crisi. !La diagnosi non va posta né negata solo sulla base dell’EEG intercritico.

Diagnostica strumentale
Se anamnesi ed esame obiettivo confermano il sospetto di crisi epilettica,
eseguire un EEG
Classificare la crisi e la sindrome epilettica
Si raccomanda l’utilizzo della Classificazione Internazionale delle Crisi e Sindromi Epilettiche
La possibilità di una Epilessia Mioclonica Giovanile dovrebbe essere presa in considerazione in adolescenti e giovani adulti che abbiano presentato una o più crisi tonico-cloniche
SI
crisi parziale crisi primariamente generalizzata
crisi non classificabile
Eseguire TC cranio o RMN encefalo
Il paziente ha meno di 25 anni di età?
È una sindrome generalizzata idiopatica?
Valutare eventuale terapia farmacologica
NO
SI
NO/INCERTO

CRISI EPILETTICA vs SINCOPESincope vasoplegica o vasovagale: In genere soggetto giovane. Spesso situazione scatenante (es. vista del sangue, temperature elevata..) e più spesso in ortostatismo. La fase prodromica è prolungata e con sintomi vegetativi. La crisi può arrestarsi in questa fase (lipotimia). La fase sincopale propriamente detta inizia con una revulsione oculare seguita da una progressiva perdita di coscienza. La caduta a terra in genere non produce trauma. La fase di incoscienza dura da pochi secondi a minuti accompagnata da pallore. Quando è prolungata si può osservare enuresi e talora irrigidimento tonico-assiale seguito da clonie irregolari (meno di 6) di ampiezza decrescente. Talvolta morsicatura della punta della lingua. Questa sintomatologia impropiamente detta “sincope convulsiva” è espressione di una sofferenza anossica ischemica delle strutture sottocorticali. TRACCIATO: scomparsa di un attività di fondo, sostituita da un'attività diffusa theta poi da un'attività delta-monomorfa. Se la crisi perdura compare un tracciato isoelettrico dove si sovrappongono gli artefatti muscolari della fase convulsiva. Test di provocazione: test di compressione oculare (più nel bambino), il “tilt test”. !Sincopi cardioplegiche: Pz più spesso di età avanzata con antecedenti che suggeriscono un'origine cardiaca (es. disturbo del ritmo, malattia coronarica, insufficienza cardiaca). Spesso nessun contesto favorente la crisi. In genere i prodromi sono assenti o brevi (es. sensazione di “testa vuota”, palpitazioni, dolore toracico..) Al sopravvenire di un'insufficienza circolatoria si ha perdita di coscienza immediata e atonia improvvisa con possibili traumatismi. Può assumere carattere “convulsivante”. !Sindrome del seno carotideo: Rara. Soprattutto nell'uomo in età matura. Fattori scatenanti costanti (es. collo della camicia stretto..).
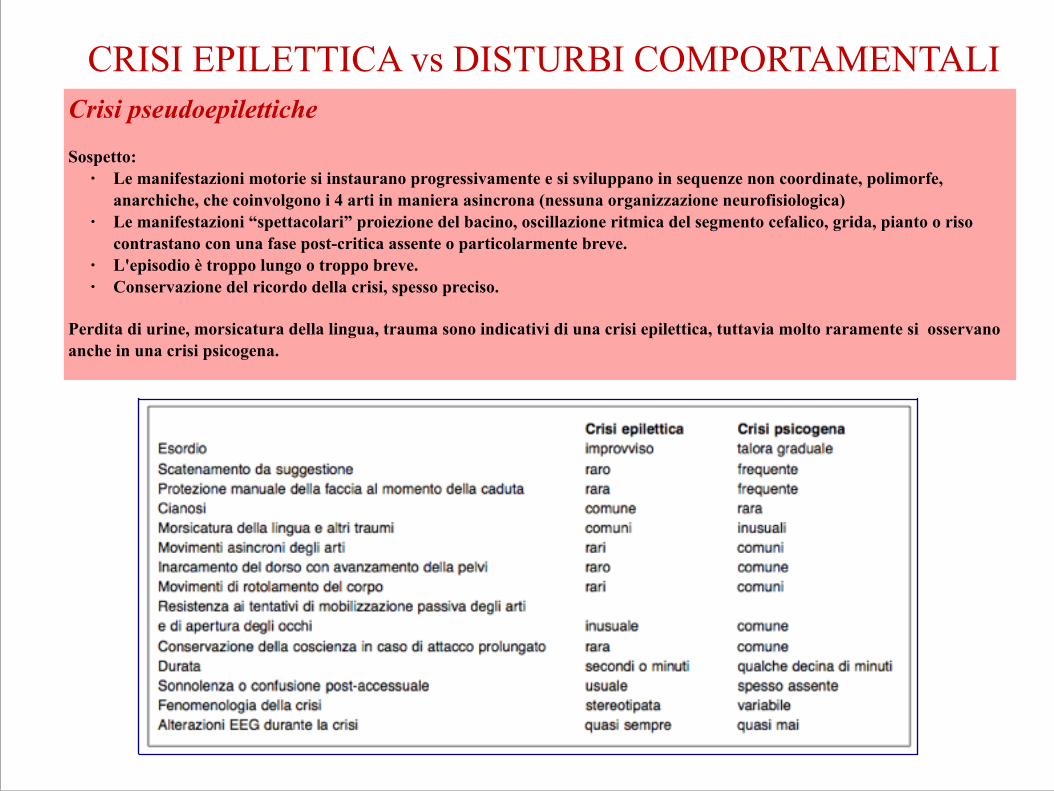
Crisi pseudoepilettiche !Sospetto:
● Le manifestazioni motorie si instaurano progressivamente e si sviluppano in sequenze non coordinate, polimorfe, anarchiche, che coinvolgono i 4 arti in maniera asincrona (nessuna organizzazione neurofisiologica)
● Le manifestazioni “spettacolari” proiezione del bacino, oscillazione ritmica del segmento cefalico, grida, pianto o riso contrastano con una fase post-critica assente o particolarmente breve.
● L'episodio è troppo lungo o troppo breve. ● Conservazione del ricordo della crisi, spesso preciso. !
Perdita di urine, morsicatura della lingua, trauma sono indicativi di una crisi epilettica, tuttavia molto raramente si osservano anche in una crisi psicogena. !!!
CRISI EPILETTICA vs DISTURBI COMPORTAMENTALI

!Attacchi di panico Episodi improvvisi di paura, coesistono polipnea, agitazione, tachicardia, sudorazione, parestesie, nausea, sensazione dolorosa toracica ed impressione di morte imminente. Possono verificarsi anche in assenza di un fattore scatenante identificabile ed in soggetti con storia psichiatrica muta. Anche se non vi è alterazione della coscienza vengono talvolta riferite sensazioni di derealizzazione e di depersonalizzazione, correlate all'intensità del disturbo ansioso che possono simulare una crisi parziale con sintomi psichici o affettivi, o una crisi parziale complessa. L'espressione clinica con sintomatologia prevalentemente vegetativa può porre il sospetto di una crisi parziale semplice con sintomi vegetativi. !Crisi simulate Sono talvolta molto difficili da differenziare dalle crisi vere. Un aspetto estremo è la Sindrome di Munchausen epilettica in cui i pazienti si sottopongono ad un grande numero di esplorazioni (anche pre-chirurgiche) in ragione di stati di male fittizzi e farmacoresistenti. Questo comportamento anomalo può verificarsi eventualmente su terza persona sindrome di Munchausen “per procura” o sindrome di Meadow una madre può affermare l'esistenza di crisi nel proprio bambino e sottoporlo a numerosi accertamenti. !Altri disturbi parossistici del comportamento Una diagnosi di epilessia errata può essere posta talvolta su comportamenti irrazionali, clastici, di collera improvvisa. Questi fenomeni non sono quasi mai di natura epilettica, ma possono complicare un'epilessia autentica.

Il trattamento viene solitamente iniziato dopo una seconda crisi tranne alcune eccezioni (disturbo cerebrale progressivo;
EEG marcatamente alterato)
TERAPIA
!1)E' preferibile iniziare con un singolo farmaco (monoterapia) La scelta del primo farmaco dipende: - dal tipo di crisi e sindrome - età e peso - tolleranza individuale del farmaco - compliance - coesistenza di altre malattie e altre terapie 2)In caso di fallimento: seconda monoterapia Introdurre un secondo farmaco (politerapia) in caso non si ottenga un completo controllo delle crisi come previsto. Complessivamente si arriva al controllo nel 70-75% delle crisi. Il 30-25% dei pazienti hanno un epilessia farmacoresistente. !E' ̀preferibile associare farmaci con meccanismo d’azione diverso. Unico criterio di efficacia: andamento della frequenza critica. ! - Il “dosaggio” plasmatico ha valore solo indicativo - La persistenza di anomalie EEG non indica fallimento terapeutico

Vecchi farmaci Fenobarbital (1912)
Primidone
Fenitoina
Carbamazepina
Etosuccimide
Valproato
Clonazepam
Diazepam
Clobazam
Lorazepam
Nitrazepam
FARMACI ANTIEPILETTICI Armamentario dei farmaci antiepilettici (AEDs)
Nuovi farmaci Gabapentin
Felbamato
Lamotrigina
Levetiracetam
Oxcarbazepina
Pregabalin
Tiagabina
Topiramato
Vigabatrin
Zonisamide
Farmaci nuovissimi
Lacosamide
Rufinamide
Stiripentolo

1. Riduzione dell’attività del canale del Na+ voltaggio-dip !2. Potenziamento della neurotrasmissione GABAergica !3. Riduzione dell’attività del canale del Ca2+ voltaggio-dip di tipo T !4. Riduzione dell’attività della neurotrasmissione glutammatergica
MECCANISMI MOLECOLARI DEI FARMACI ANTIEPILETTICI


















