
DIPARTIMENTO DI CHIMICA E CHIMICA INDUSTRIALE · che segue la regola EAN, il numero di elettroni di...
79
UNIVERSITÀ DI PISA DIPARTIMENTO DI CHIMICA E CHIMICA INDUSTRIALE CORSO DI LAUREA MAGISTRALE IN CHIMICA (CLASSE LM-54 SCIENZE CHIMICHE) CURRICULUM INORGANICO SINTESI E REATTIVITÀ DI CLUSTER CARBONILICI DI FERRO Candidato: Relatore: Nicola Salvo Prof. Piero Leoni Controrelatore: Dr. Tiziana Funaioli Tesi di Laurea Magistrale Anno Accademico 2013/2014
Transcript of DIPARTIMENTO DI CHIMICA E CHIMICA INDUSTRIALE · che segue la regola EAN, il numero di elettroni di...

UNIVERSITÀ DI PISA
DIPARTIMENTO DI CHIMICA E CHIMICA INDUSTRIALE
CORSO DI LAUREA MAGISTRALE IN CHIMICA
(CLASSE LM-54 SCIENZE CHIMICHE)
CURRICULUM INORGANICO
SINTESI E REATTIVITÀ DI
CLUSTER CARBONILICI DI FERRO
Candidato: Relatore:
Nicola Salvo Prof. Piero Leoni
Controrelatore:
Dr. Tiziana Funaioli
Tesi di Laurea Magistrale
Anno Accademico 2013/2014

i
INDICE
RIASSUNTO .......................................................... iv
1 INTRODUZIONE ................................................ 1
1.1 Cos’è un cluster ................................................................................ 1
1.1.1 Strutture ed elettroni di valenza in un cluster ...................................... 3
1.2 Polimeri 2.0 ..................................................................................... 8
1.2.1 Cluster nella catena principale di un polimero ..................................... 9
1.2.2 Cluster connessi alla catena principale di un polimero ............................ 111
1.2.3 Cluster nella catena laterale di un polimero ...................................... 12
1.2.4 Catene lineari di metalli e strutture dendrimeriche ............................ 113
1.3 Cluster di platino ........................................................................... 16
1.3.1 Polimeri con cluster di platino ......................................................... 18
1.4 Perché i cluster di ferro ......................................................... 202020
1.4.1 Cluster carbonilici di ferro ............................................................ 221
2 SCOPO DELLA TESI ...................................... 24
3 RISULTATI E DISCUSSIONE ...................... 25
3.1 Sintesi e reattività dei cluster carbonilici di ferro ............. 25222525
3.1.1 Sintesi, caratterizzazione e reattività del cluster Fe5C(CO)15 (1) .......... 25
3.1.2 Sintesi, caratterizzazione e reattività del cluster
Fe5C(CO)14(PHtBu2) (2) ......................................................................... 128
3.1.3 Sintesi e caratterizzazione del cluster [Et4N]2[Fe6C(CO)16] (3) .......... 331

ii
3.1.4 Sintesi e caratterizzazione del cluster [Et4N]2[Fe5C(CO)14] (5) ........... 132
3.2 Sintesi di sali contenenti cluster nel catione e nell’anione 25222533
3.3 Sintesi di cluster bimetallici .............................................. 25222536
3.3.1 Sintesi, caratterizzazione e reattività di
[Et4N]2[Fe5C(CO)14(CuCl)] (6) .............................................................. 137
3.3.2 Sintesi del cluster [Et4N]2[{Fe6C(CO)16}2(Hg)] (7) ........................... 39
3.4 Misure elettrochimiche 40
3.4.1 Misure di voltammetria ciclica ....................................................... 40
3.4.2 Esperimenti di spettroelettrochimica ................................................. 45
3.5 Riduzione chimica e riossidazione di Fe5C(CO)14(PHtBu2) 48
4 CONCLUSIONI ................................................. 50
5 PARTE SPERIMENTALE .............................. 52
5.1 Generalità 52
5.2 Solventi e reagenti 52
5.3 Misure chimico-fisiche 53
5.3.1 Misure elettrochimiche ................................................................. 53
5.4 Procedure sperimentali 55
5.4.1 Sintesi di Fe5C(CO)15 (1) ................................................................ 55
5.4.2 Sintesi di Fe5C(CO)14(PHtBu2) (2) .................................................. 56
5.4.3 Sintesi di [Et4N]2[Fe6C(CO)16] (3) ................................................... 56
5.4.4 Preparazione del sale misto
[Pt3(µ-PtBu2)3(CO)3][Et4N][Fe6C(CO)16] (4) ............................................. 57
5.4.5 Sintesi di [Et4N]2[Fe5C(CO)14] (5) ................................................... 57
5.4.6 Sintesi di [Et4N]2[Fe5C(CO)14(CuCl)] (6) ......................................... 57
5.4.7 Sintesi di [Et4N]2[{Fe6C(CO)16}2(Hg)] (7) ....................................... 58
5.4.8 Riduzione di Fe5C(CO)14(PHtBu2) (2) e successiva riossidazione .......... 58

iii
APPENDICE I Voltammetria ciclica ............... 60
APPENDICE II Spettroelettrochimica ............. 65
BIBLIOGRAFIA .................................................... 68

iv
RIASSUNTO
In questo lavoro di tesi è stata effettuata un’indagine sulla sintesi e la reattività di
cluster carbonilici di ferro, in particolare di quelli con un atomo di carbonio
interstiziale data la loro maggiore stabilità strutturale. Ciò allo scopo di individuare
possibili sintoni da utilizzare nella costruzione di strutture macromolecolari, in
analogia con quanto fatto sui cluster di platino studiati nel laboratorio in cui è stato
svolto il lavoro di tesi. Utilizzando procedure note in letteratura [49a, 58a] sono stati
sintetizzati due cluster precursori, [Fe5C(CO)15] (1) e [Fe6C(CO)16]2- (3), che sono stati
poi utilizzati come reagenti di partenza sia per la sintesi dei nuovi composti
Fe5C(CO)14(PHtBu2) (2) e [Pt3(µ-PtBu2)3(CO)3][Et4N][Fe6C(CO)16] (4) sia per la
preparazione dei già noti [Et4N]2[Fe5C(CO)14] (5) [49a], [Et4N]2[Fe5C(CO)14(CuCl)]
(6) [53] e [Et4N]2[{Fe6C(CO)16}2(Hg)] (7) [55a]. Dalla reazione di 1 con la fosfina
secondaria PHtBu2 è stato ottenuto il cluster 2 per sostituzione di un legante
carbonilico. La scelta di usare una fosfina secondaria come PHtBu2 va legata alla
possibilità di intervenire con ulteriori reazioni sul centro reattivo costituito dal legame
P-H: le prove di deprotonazione della fosfina coordinata sono state effettuate nel
tentativo di ottenere un fosfuro nucleofilo capace di reagire con un cluster elettrofilico
al fine di aumentare la nuclearità dei sistemi studiati. Oltre a ciò, il cluster 2 è stato
sottoposto ad indagini di tipo elettrochimico che hanno messo in evidenza la possibilità
di dissociare reversibilmente la fosfina coordinata all’unità {Fe5C(CO)14} al variare
del potenziale applicato.
A partire da 3 e [Pt3(µ-PtBu2)3(CO)3]CF3SO3 è stato ottenuto il sale organometallico
4 contenente due diverse unità cluster. L’importanza di questa classe di composti,
piuttosto rari, è legata sia al loro potenziale uso come intermedi per la sintesi di cluster
a nuclearità più elevata sia alla possibilità di avere trasferimento elettronico fra le unità
cluster che costituiscono il sale. Le indagini voltammetriche effettuate sul sale 4 hanno
però messo in evidenza l’assenza di scambio elettronico fra le due unità cluster che lo
costituiscono.
La necessità di disporre di specie che possono dare reazioni di sostituzione di un
legante in modo ben definito ha sospinto le indagini nei confronti dei cluster

v
bimetallici, soprattutto quelle riguardanti il cluster [Et4N]2[Fe5C(CO)14(CuCl)] (6), nel
quale il rame mantiene il suo stato di ossidazione formale +1, allo scopo di ottenere
diversi altri derivati attraverso reazioni di sostituzione del legante cloruro. Le prove di
sintesi di derivati alchinilici del composto 6 non hanno però portato alla sostituzione
del legante cloruro coordinato al rame con l’alchinile ma, piuttosto, alla
decomposizione del cluster.

1
1
INTRODUZIONE
1.1 Cos’è un cluster
Nel 1964 F. A. Cotton scriveva: “Il termine cluster sembra quello più adatto a definire
un gruppo finito di atomi di un metallo, i quali sono tenuti insieme prevalentemente, o
almeno in misura significativa, mediante legami metallo-metallo, anche se alcuni
atomi non metallici possono essere intimamente associati con il cluster” [1].
Tale termine andò a sostituire quelli di Staphylonuclear e Cage usati in precedenza.
Un cluster metallico può quindi essere definito, in maniera più rigorosa, come una
specie costituita da un numero finito di atomi metallici (almeno 3) tenuti insieme fra
loro attraverso legami metallo-metallo, la cui presenza è stata indiscutibilmente
stabilita; oltre all’unità polimetallica un cluster metallico molecolare può contenere un
certo numero di leganti coordinati, terminalmente o a ponte, ai centri metallici, i quali
possono essere localizzati, a seconda della forma e della dimensione dell’unità
polimetallica, sia sulla superficie che all’interno della stessa [2, 3]. Il lavoro di Cotton
pubblicato nel 1964 rappresenta una pietra miliare nell’ambito della chimica dei
cluster, che da allora e dai primi lavori pubblicati dallo stesso Cotton sui dimeri e
trimeri di Re(III), in Fig. 1.1, in cui sono presenti legami multipli metallo-metallo [4],
è cresciuta in modo esponenziale, grazie anche al continuo miglioramento delle
tecniche di indagine (come la diffrazione di raggi X) indispensabili per la
determinazione delle strutture e l’individuazione dei legami fra i centri metallici.
Figura 1.1 Stutture di [Re2Cl8]2- e [Re3Cl12]3-.

2
I cluster dei metalli di transizione possono essere suddivisi in cluster organometallici
e cluster inorganici [3]. Alla prima classe appartengono quelli formati da metalli in
basso stato di ossidazione (≤ 2) con leganti soft π-acidi (in genere CO) aventi nuclearità
variabile, a volte anche molto elevata, ed in presenza di legami metallo-metallo di
ordine basso (unitario o frazionario); inoltre, nei cluster con un numero elevato di
atomi di metallo, appartenenti alla classe dei sistemi organometallici, è frequente la
presenza di atomi interstiziali che rendono la struttura più stabile. Alla classe dei
cluster inorganici appartengono specie caratterizzate dalla presenza di atomi di metallo
in stato di ossidazione medio e leganti σ-donatori (spesso anche π-donatori) come
alogenuri, alcossidi, carbossilati e ammidi; per tali cluster la nuclearità è generalmente
bassa mentre è elevata la probabilità di trovare legami multipli metallo-metallo [3, 5].

3
1.1.1 Strutture ed elettroni di valenza in un cluster
Gli atomi dei metalli tendono ad aggregarsi secondo differenti tipologie strutturali
formando ad esempio poliedri regolari, come il tetraedro, la bipiramide trigonale,
l’ottaedro, o strutture meno simmetriche ottenute dai deltaedri regolari (poliedri con
facce triangolari) per rottura di legami metallo-metallo (Fig. 1.2). Mentre i cluster a
bassa nuclearità (M ≤ 6) si presentano con strutture aventi elevata simmetria, quelli a
nuclearità più alta mostrano strutture che possono essere derivate da quelle più
semplici per aggiunta di atomi metallici a ponte sugli spigoli o sulle facce triangolari
[2].
Figura 1.2 Geometrie assunte dai cluster con nuclearità Mx [2].

4
Strutture pseudo-sferiche (cubiche, icosaedriche) divengono invece più comuni solo
in presenza di atomi interstiziali. Cluster con atomi interstiziali possono essere formati
anche quando la nuclearità è bassa: in questi casi all’interno delle cavità vengono
ospitati piccoli atomi (H, C, N); nei cluster a nuclearità più alta, in particolare con M
≥ 8, si possono invece avere atomi interstiziali più pesanti dei gruppi principali, come
S, Se, P, As, (Fig. 1.3) o addirittura atomi metallici (Ru, Os, Co, Rh, Ni, Pd) nei casi
in cui M ≥ 11[2].
La disposizione dei centri metallici spesso richiama l’impaccamento compatto (hcp,
fcc) che viene osservato nei metalli e ciò è più frequente laddove la nuclearità del
cluster è elevata, anche se le geometrie assunte dai cluster aventi nuclearità minore
(triangolare, tetraedrica, ottaedrica) si possono considerare come parti di un
impaccamento esagonale compatto, come riassunto in Fig. 1.4 [2].
Figura 1.3 [Rh9P(CO)21]2-.
Figura 1.4 Confronto tra la struttura compatta di una superficie metallica e la gabbia metallica di un cluster.

5
La struttura di un cluster può essere prevista correlando in modo opportuno nuclearità
e numero di elettroni di valenza (NEV). Per quanto riguarda il numero di elettroni di
valenza, va detto che soltanto i cluster di piccole dimensioni (M ≤ 5) seguono la regola
dei 18 elettroni o del numero atomico effettivo (EAN), con diverse eccezioni, mentre i
cluster a nuclearità maggiore la rispettano solo di rado [3, 6]. Va ricordato che la regola
dei 18 elettroni, elaborata nel tentativo di interpretare la struttura dei complessi
metallocarbonilici da I. Langmuir nel 1921 [7], si basa sul fatto che un centro metallico
consta di nove orbitali di frontiera (un orbitale s, tre p e cinque d) nei quali possono
essere ospitate 9 coppie di elettroni derivanti sia dal metallo che dai leganti ad esso
coordinati e che consentono il raggiungimento di una configurazione a guscio chiuso
stabile, analoga a quella del gas nobile corrispondente. Di conseguenza in un cluster
che segue la regola EAN, il numero di elettroni di valenza è correlato alla nuclearità
(x) e al numero di legami metallo-metallo (y) mediante la seguente equazione [3]:
NEV = 18x – 2y (1.1).
Come già detto, quando la nuclearità è elevata i cluster non seguono più la regola dei
18 elettroni e di conseguenza previsioni sulla struttura a partire dal numero di elettroni
di valenza possono essere fatte ricorrendo ad altre regole, le più generali delle quali
sono le regole di Wade-Mingos conosciute anche come teoria PSEP (Polyhedral
Skeletal Electron Pair) che adattano le regole enunciate per i composti polinucleari del
boro (borani e carborani) ai cluster dei metalli di transizione e post-transizione aventi
geometrie pseudo-sferiche (closo) o derivate da esse per allontanamento di un vertice
(nido), o di due vertici (aracno) [6, 8]. Anche il boro, infatti, non rispetta sempre la
regola dell’ottetto (analoga a quella dei 18 elettroni valida per gli elementi di
transizione) e si ritrova spesso con un numero di elettroni di valenza inferiore rispetto
a quelli attesi: nei borani, ad esempio, la lacuna elettronica intorno agli atomi di boro
viene compensata mediante la formazione di legami multicentrici, come quelli a 3
centri e 2 elettroni, che comportano un aumento dell’energia di alcuni orbitali
molecolari (MO) leganti, i quali divengono MO di non legame o di antilegame. In
questo modo si ha una riduzione degli MO leganti con conseguente formazione di
aggregati stabili, essendo tutti gli orbitali leganti occupati, anche in presenza di un
NEV inferiore a quello necessario a dare legami localizzati 2c-ne- (con n = 2, 4, 6).

6
Appare evidente che nei sistemi in cui sono presenti legami multicentrici il singolo
legame elemento-elemento sarà in genere di ordine inferiore ad 1 [3].
Nei cluster dati dai metalli di transizione ciascun centro metallico ha a disposizione
nove orbitali di valenza, sei dei quali sono impiegati per la coordinazione dei leganti;
i tre orbitali restanti sono utilizzati in modo da generare n+1 MO dedicati alla
costruzione dello scheletro di un cluster MnLx closo. Gli MO di scheletro diventano
n+2 o n+3, se la struttura è rispettivamente nido o aracno. In totale si hanno 7n+1,
7n+2 o 7n+3 MO, che corrispondono a 14n+2, 14n+4 o 14n+6 elettroni di valenza per
le strutture closo, nido o aracno (Fig. 1.5). Tali relazioni valgono per cluster di metalli
di transizione o di elementi dei gruppi principali con geometrie pseudosferiche o
derivate.
Dal calcolo del NEV, ottenuto sommando gli elettroni di valenza dei centri metallici
con quelli forniti dai leganti e tenendo conto della eventuale carica totale del cluster, è
in genere possibile fare una previsione ragionevole sulla struttura dello stesso. In
Tabella 1.1 si riporta la corrispondenza sussistente tra le strutture più comuni osservate
nell’ambito della chimica dei cluster e il NEV.
Figura 1.5 Esempi di strutture closo (a, e), nido (b, f) e aracno (c, d, g) di cluster metallo carbonilici
aventi un NEV rispettivamente di 14n+2, 14n+4 e 14n+6 (n è il numero di vertici).

7
Per specie meno simmetriche sono state elaborate altre relazioni NEV-struttura, come
ad esempio le regole di capping o quelle per poliedri condensati. Le regole di Wade-
Mingos sono valide fino a nuclearità di poco superiori a 12, mentre per cluster di
dimensioni maggiori vi sono altre relazioni che legano il numero di elettroni di valenza
del cluster alla sua struttura, che tuttavia hanno un’affidabilità tanto più bassa quanto
più alta è la nuclearità [9]. All’aumentare del numero di centri metallici, infatti, gli
orbitali molecolari diventano sempre più numerosi e ravvicinati in energia e ciò fa sì
che un dato cluster risulti stabile con un numero variabile di elettroni: questa possibilità
rende tali cluster capaci di funzionare da riserva di elettroni, ossia come capacitori
molecolari [10, 11]. Si può quindi presumere un graduale passaggio da un
comportamento isolante a quello tipico di un semiconduttore e, oltre una certa soglia,
a quello di conduttore elettrico. L’interesse nei confronti di queste proprietà e le
potenziali applicazioni che ne possono conseguire nell’ambito dell’elettronica
molecolare [12] hanno contribuito negli ultimi anni alla ricerca e alla messa a punto di
nuove metodologie sintetiche per la sintesi di cluster a nuclearità sempre più elevata,
che sono solitamente chiamati “cluster giganti”. Preparati principalmente dai gruppi
di ricerca di Dahl [13], Longoni [10] e Schnöckel [14] e caratterizzati per via
diffrattometrica, oggi tra i sistemi a più alta nuclearità vanno citati
PtPd164(CO)72(PPh3)20, [Al77{N(SiMe3)2}20]2- e [Ga84{N(SiMe3)2}20]
3-. L’interesse nei
confronti di questi sistemi di grandi dimensioni va altresì legato a quello rivolto negli
Geometrie NEV Esempi
Triangolo 48 Os3(CO)12
Tetraedro 60 Rh4(CO)12
Quadrato planare 64 Pt4(MeCOO)8
Bipiramide trigonale 72 Os5(CO)16
Piramide quadrata 74 Fe5C(CO)15
Ottaedro 86 Rh6(CO)16
Prisma trigonale 90 [Rh6C(CO)15]2-
Antiprisma quadrato 114 [Co8C(CO)18]2-
Cubo 120 Ni8(PPh)6(CO)8
Icosaedro 170 [Rh12Sb(CO)27]3-
Tabella 1.1 NEV e corrispondenti geometrie [2].

8
ultimi tempi alle nanoparticelle metalliche (particelle polidisperse con composizione
e struttura non perfettamente definite) che, al contrario dei cluster molecolari, stanno
diventando sempre più piccole: si pensa, infatti, che proprio dallo studio delle proprietà
e delle caratteristiche dei cluster giganti si possa avere una migliore comprensione
delle proprietà delle nanoparticelle, come ad esempio gli effetti quantici correlati alle
loro dimensioni [15]. Tra le altre motivazioni che hanno spinto, nel corso degli anni,
la ricerca sui cluster molecolari va ricordata, infine, la ricerca delle eventuali analogie
sussistenti tra le strutture e la reattività dei cluster e quella delle superfici metalliche
[16], questione di grande rilievo al fine della razionalizzazione e della comprensione
dell’attività catalitica svolta dalle superfici di molti metalli.
1.2 Polimeri 2.0
Il progresso tecnologico di ogni società è indissolubilmente legato ai materiali a
disposizione della società stessa, ed in effetti, gli storici hanno sfruttato la nascita e lo
sviluppo di nuovi materiali come riferimento al fine di stabilire un punto fermo di
datazione nel percorso storico della civilizzazione dell’uomo. Tra i materiali che, a
partire dagli anni ’30, hanno avuto un forte sviluppo ed una diffusione quasi capillare
a tutti i livelli della società vi sono i polimeri: la versatilità di questi composti, unita
all’enorme disponibilità ed al basso costo delle materie prime (essenzialmente derivati
del petrolio), ha permesso la produzione di materiali dotati di un ventaglio di proprietà
impossibile da ottenere usando fonti primarie come il legno, i metalli o le
ceramiche[17]. Tale ampio spettro di caratteristiche è divenuto ancora più ampio da
quando la scienza dei polimeri si è accostata ai metalli di transizione: dai primi
polimeri organometallici, come ad esempio il poli(vinil)ferrocene descritto per la
prima volta nel 1955 [18] o il poliferrocenilene preparato nei primi anni ’80 (Fig 1.6)
e i suoi vari derivati [19], si giunge ai più complessi e meno noti composti polimerici
contenenti cluster metallici come unità strutturali.
Figura 1.6 Poliferrocenilene [18].

9
La presenza di metalli di transizione lungo la catena di un polimero consente, ad
esempio, il controllo redox della dimensione e della forma di un oggetto macroscopico
[20], la formazione di legami selettivi con piccole molecole, ioni metallici o
biomolecole [21], la generazione di nuovo materiale con attività catalitica [22] e la
generazione di nuove proprietà fotofisiche [23]. In particolare, anche i polimeri
contenenti cluster, sebbene ancora poco conosciuti, mostrano eccellenti potenzialità
nella preparazione di materiali funzionali con varie caratteristiche e sono attualmente
oggetto di studio. Similmente a quanto accade con i polimeri “convenzionali”, anche
per queste macromolecole contenenti cluster metallici nella loro struttura sono
possibili diverse tipologie di architettura polimerica che possono essere
sostanzialmente distinte in base alle differenti posizioni rispetto allo scheletro
principale del polimero a cui i cluster metallici sono legati, in analogia con quanto fatto
per i polimeri organometallici [24] (Fig. 1.7).
Nel caso A) di Figura 7, il cluster fa parte della catena principale e la sua presenza è
indispensabile per l’esistenza del polimero; negli altri due casi, invece, l’eventuale
dissociazione del cluster non provoca la distruzione della struttura polimerica, essendo
questo o direttamente legato alla catena principale del polimero (caso B), o legato ad
essa attraverso una catena laterale (caso C).
1.2.1 Cluster nella catena principale di un polimero
I primi polimeri contenenti cluster nella catena principale (Fig. 1.8) sono stati
sintetizzati da Puddephatt et al. nel 1994 [25]: tali macromolecole sono state ottenute
Figura 1.7 Strutture possibili di un polimero contenente un cluster metallico: A) cluster nella catena
principale; B) cluster legato in maniera diretta alla catena principale; C) cluster in catena laterale.

10
facendo reagire il cluster triangolare cationico di platino [Pt3(µ3-CO)(µ-dppm)3]2+ con
diisocianuri, CNC6R4NC (R = H, Me), a dare complessi polimerici del tipo [Pt3(µ-
dppm)3(µ-CNC6R4NC)]n2n+, nei quali i diisocianuri fanno da ponte tra le unità Pt3.
Questa classe di polimeri contenenti cluster triangolari di platino o altre unità Mn
connesse da leganti isocianuro, o anche da leganti al fosforo o all’azoto, mostra però
una scarsa solubilità nei solventi non polari e di conseguenza la loro caratterizzazione
spesso è risultata difficoltosa. Tra i pochi solventi in cui questi composti risultano
solubili si annoverano l’acetone o il dimetilsolfossido (DMSO), nei quali però si può
avere la frammentazione del polimero stesso. Al contrario, Johnson et al. hanno
sintetizzato per primi dei polimeri aventi unità di cluster carbonilici all’interno della
catena principale [26], caratteristica che ha reso tali macromolecole più solubili nei
solventi poco polari e che di conseguenza ne ha consentito una migliore e più facile
caratterizzazione. [Ru6C(CO)15(Ph2P-CC-PPh2)]n è stato ottenuto per reazione di
[Ru6C(CO)17] con la difosfina Ph2P-CC-PPh2 in tetraidrofurano (THF) in condizioni
di riflusso, con un grado medio di polimerizzazione di circa 1000 [26]. Il lavoro di
Humprey et al. rappresenta, invece, un esempio di polimeri aventi lungo la catena
principale cluster bimetallici: nello specifico, si tratta di una serie di oligouretani
contenenti unità dimolibdeno-diiridio (Mo2Ir2) nello scheletro dell’oligomero ottenuti
mediante policondensazione a stadi (Fig. 1.9) [27].
Figura 1.8 Polimeri sintetizzati da Puddephatt et al. nel 1994 [25].

11
1.2.2 Cluster connessi alla catena principale di un polimero
Come già anticipato nella sezione 1.2, vi sono esempi di polimeri nei quali il cluster
metallico non fa parte della catena polimerica principale ma lo si ritrova direttamente
connesso ad essa (caso B, Fig. 1.7). Un esempio è dato dal lavoro di Han e collaboratori
che nel 2003 riportarono la sintesi e la caratterizzazione di un copolimero azulene-
tiofene che presenta unità nelle quali un cluster carbonilico di rutenio è coordinato alla
porzione azulenica: la sintesi, come riassunto in Fig. 1.10, è stata effettuata mediante
polimerizzazione ossidativa di derivati dell’1,3-di(tiofen-2-il)azulene con differenti
catene laterali R (R = C10H21 e OC12H25) in presenza di FeCl3 e successivo riflusso,
dei polimeri π-coniugati ottenuti, in xilene in presenza del cluster carbonilico di rutenio
[Ru3(CO)12] [28].
Figura 1.10 Schema di sintesi di 4 differenti polimeri con il cluster
carbonilico di rutenio legato alla porzione azulenica [28].
Figura 1.9 Oligouretani sintetizzati da Humprey et al. nel 2001 [27].

12
Tali sistemi si sono rivelati interessanti poiché la composizione del cluster carbonilico
di rutenio influenza le caratteristiche della macromolecola finale, permettendo così di
modularne le proprietà elettroniche, ottiche e morfologiche [28].
1.2.3 Cluster nella catena laterale di un polimero
Cluster di metalli di transizione possono essere legati alle catene laterali di un polimero
(caso C, Fig. 1.7): in questi casi, il cluster può legarsi a polimeri funzionalizzati
preformati oppure è possibile ottenere il polimero per oligomerizzazione di un cluster
che contiene un doppio legame nella regione periferica di uno dei leganti. In genere, il
cluster è coordinato al polimero mediante legame dativo con atomi neutri donatori
come N, P, O. Tra i diversi esempi di polimeri o metallopolimeri di questo tipo [29] si
segnala il lavoro di Zheng et al. in cui viene riportata la sintesi di un copolimero di
stirene con [Re6(µ3-Se)8(PEt3)5(4-vinilpiridina)](SbF6)2, in accordo con lo schema
riportato in Fig. 1.11 [30].
Questo copolimero organico-inorganico contenente un metallo di transizione ed un
calcogeno si presenta con un basso indice di polidispersione ed un alto peso molecolare
medio [30]. Metallopolimeri con cluster in catena laterale sono quelli preparati da
Chan et al.: in questo caso vengono legati, alle catene laterali di poliferrocenilsilani
con sostituenti acetilenici, cluster di Mo, Ni e Co, come di seguito riportato in Fig.
1.12 [31].
Figura 1.11 Copolimerizzazione con stirene in acetonitrile in presenza di
2,2´-azo-bis-isobutirrilnitrile (AIBN) come iniziatore [30].

13
Gli studi effettuati suggeriscono che tali polimeri altamente metallizzati possono
essere utilizzati per fabbricare nanostrutture contenenti ferro, interessanti per le
proprietà magnetiche e catalitiche, mentre, più in generale, composti di questo tipo
possono risultare utili sia nell’ambito della scienza dei materiali che in ambito
catalitico [31].
1.2.4 Catene lineari di metalli e strutture dendrimeriche
Polimeri con legami metallo-metallo lungo la catena principale sono noti da più di 40
anni: il primo polimero metallico a struttura lineare, infatti, è stato sintetizzato da
Krogman nel 1969 (Fig. 1.13) [32].
Figura 1.13 Allo stato solido composti di Pt(II) con geometria quadrato
planare, di formula generale M2[Pt(CN)4], detti Sali di Krogman,
formano catene infinite allineando i centri metallici lungo l’asse z [32].
Figura 1.12 Schema di reazione del metallopolimero con il corrispondente cluster in toluene [31].

14
Questi composti hanno ricevuto negli ultimi anni una certa attenzione poiché da essi
si è cercato di sviluppare i cosiddetti fili molecolari (o nanofili molecolari) in grado di
condurre la corrente elettrica. Tali oggetti molecolari sono schematicamente costituiti
da una catena lineare di atomi di metallo circondata da un isolante (il legante) organico
che, essendo direttamente legato agli atomi metallici, contribuisce a determinare le
proprietà di conduzione complessive del filo. Esempi di sistemi di questo tipo
comprendono materiali polimerici come Mo6S9-xIx (4 < x < 6) in Fig. 1.14 [33] o come
Li2Mo6Se6 [33c].
Come prototipi di EMACs (extended metal atom chains) Cotton et al. hanno studiato
complessi trinucleari lineari del tipo M3(dpa)4L2 (con dpa = 2,2´-dipiridilammide) che,
a seconda dello stato di ossidazione del metallo, possono formare catene lineari
mediante la formazione di legami M-M [34]. Vanno citati anche i sistemi a base di
palladio, nei quali catene lineari monodimensionali di Pd sono ottenute facendo reagire
sistemi polienici coniugati con [Pd(CH3CN)4][BF4]2 e [Pd2(dba)3] (Fig. 1.15) [35].
Figura 1.14 Immagini SEM di fili molecolari di Mo6S9-xIx (4 < x < 6) [33].
Figura 1.15 A sinistra, schema di sintesi di un sistema Pd4; a destra, schema di sintesi generale [35].

15
I dendrimeri sono una classe di composti macromolecolari polimerici aventi strutture
iper-ramificate di forma globulare. Il termine dendrimero deriva dal greco δένδρον che
significa albero, in analogia con la sua struttura. In generale le proprietà dei dendrimeri
sono determinate dai gruppi funzionali presenti sulla superficie molecolare: ad
esempio un dendrimero può essere idrosolubile se i suoi gruppi esterni sono
idrosolubili. In letteratura vi sono vari esempi di cluster di metalli di transizione
coordinati alle catene esterne di dendrimeri organici [36]: questa tipologia di composti
può apportare vantaggi unici, ad esempio, nella preparazione di particelle
monodisperse con strutture molecolari e funzionalità di superficie ben definite. Sono
noti anche dendrimeri in cui il cluster metallico si trova al centro della struttura
piuttosto che sulla sua superficie: un esempio è dato dal dendrimero preparato da Méry
et al., nel quale un cluster esanucleare, a geometria ottaedrica, di molibdeno è stato
fatto reagire con piridine dendroniche, secondo lo schema riportato in Fig. 1.16 [37].
Figura 1.16 Schema di sintesi del dendrimero con il core costituito dal cluster Mo6 sintetizzato da
Méry et al. [37].

16
1.3 Cluster di platino
I cluster di metalli di transizione sono composti interessanti per svariati motivi, tra cui
la possibilità di fare da trait d’union tra il semplice complesso metallico e i colloidi, le
particelle e le superfici metalliche. L’interesse nei confronti dei cluster è stato fin dai
primi studi ulteriormente sospinto dal riconoscimento delle loro proprietà catalitiche e
dalle eventuali nuove possibilità che ciò poteva aprire. In particolare, parecchio
studiati, almeno in un primo tempo, sono stati i cluster trinucleari [38] come modello
più semplice di una superficie metallica: da citare in tal senso vi è la famiglia M3(CO)12
(M = Fe, Ru, Os); questi cluster sono elettronicamente saturi con un adeguato numero
di elettroni condivisi da ciascun metallo in modo da raggiungere la configurazione a
guscio chiuso a 18 elettroni di valenza. Relativamente alle specie trinucleari, anche il
platino ha una forte tendenza a formare aggregati basati sull’unità triangolare: in questa
struttura i tre atomi di platino e gli atomi donatori dei leganti L ed L´ sono
approssimativamente coplanari, come visibile in Fig. 1.17.
Tra gli aggregati basati sull’unità triangolare si possono annoverare come esempio
tutte le specie colonnari con stechiometria [Pt3(CO)6]n2- (n = 2-5), che sono costruite
mediante sovrapposizione delle unità trinucleari planari Pt3(CO)6 (Fig. 1.18) [39].
Figura 1.18 Strutture di [Pt(CO)3(µ-CO)3]32- e [Pt(CO)3(µ-CO)3]5
2- [39b].
Figura 1.17 Struttura di un cluster trinucleare di platino.

17
In queste strutture le distanze Pt-Pt all’interno di ogni unità Pt3 sono molto più brevi
di quelle Pt-Pt fra due triangoli adiacenti; la sovrapposizione fra le unità triangolari,
inoltre, si verifica con i piani Pt3 quasi paralleli tra di loro, con un piccolo slittamento
laterale a favorire una disposizione elicoidale di questa sorta di cavo molecolare [39].
Sono stati descritti anche composti simili del nichel, sebbene rispetto al platino la
nuclearità massima risulta più piccola [39]. I cluster trinucleari di platino, e non solo
quelli trinucleari, costituiscono dei sintoni efficienti per la preparazione di strutture più
estese: in particolare, nel corso degli ultimi anni, nel laboratorio in cui è stato svolto il
presente lavoro di tesi, sono stati preparati e studiati i cluster [Pt3(µ-
PtBu2)3(CO)3]CF3SO3 a 44 e- e [Pt6(µ-PtBu2)4(CO)6](CF3SO3)2 a 82 e-. Entrambi questi
cluster sono stati preparati a partire dal precursore triangolare Pt3(µ-PtBu2)3(CO)2H
(Fig. 1.19), il quale a sua volta è stato sintetizzato da K2PtCl4 [40].
I suddetti cluster, le cui strutture allo stato solido ed in soluzione sono state determinate
mediante studi cristallografici e spettroscopici, risultano adatti per la costruzione di
strutture macromolecolari in virtù del fatto che essi possono essere preparati con rese
e purezza soddisfacenti, sono stabili all’aria e si mostrano termicamente robusti, hanno
un core polinucleare notevolmente stabile che rimane invariato nelle condizioni di
reazione necessarie a costruire le macromolecole e soprattutto presentano siti reattivi
Figura 1.19 Schema di sintesi di [Pt3(µ-PtBu2)3(CO)3]+ e di [Pt6(µ-PtBu2)4(CO)6]2+: la protonazione
di 1 con una quantità equimolare di acido triflico sotto atmosfera di CO porta al cluster trinucleare 2,
che si presenta come un solido di colore verde; la stessa procedura in presenza di un eccesso di acido
triflico porta al cluster esanucleare 3, che si presenta come un solido cristallino rosso [40].

18
nella giusta posizione e con la giusta reattività. A sostegno del potenziale sintetico di
questi precursori si riportano in Fig. 1.20 gli schemi sintetici di un derivato lineare e
di un dendrimero, con una struttura quasi planare, contenenti unità Pt6 e/o Pt3 connesse
mediante spaziatori alchinilici coniugati [41].
H H
Cl
CuIEt2NH
H
ClCl
CuIEt2NH
H
H
H
Cl
CuIEt2NH
27
28
16
(25)
(10)25
Scheme 9
1.3.1 Polimeri con cluster di platino
Sebbene cluster esanucleari veri e propri, nei quali i sei atomi di platino formano un
unico poliedro centrale, sono molti rari, nonostante siano noti molti cluster triangolari
che in teoria possono dimerizzare in molti modi, cluster esanucleari, derivati da [OC-
{Pt6}-CO](CF3SO3)2 (con {Pt6} = {Pt6(µ-PtBu2)4(CO)4}), sono stati utilizzati per
sintetizzare i primi polimeri σ-alchinilici contenenti cluster di metalli di transizione
[42]. I derivati alchinilici e bis-alchinilici dei cluster precursori di questi polimeri sono
stati preparati a partire da [Cl-{Pt6}-Cl] per reazione con il legante alchinilico
desiderato, in condizioni tipo Sonogashira, come nel caso riportato in Fig. 1.21 [42].
Figura 1.20 Schema di sintesi di due derivati contenenti le unità Pt6 e Pt3 [41].
Figura 1.21 Schema di sintesi di un polimero con reazione in condizioni di tipo
Sonogashira, contenente l’unità Pt6 [42].

19
Questa tipologia di macromolecole può avere potenzialmente importanti proprietà
fotofisiche ed elettroniche. Riguardo a quest’ultimo punto, su derivati dei precursori
di questi polimeri, sono stati effettuati studi sui fenomeni di trasferimento di carica.
Ad esempio, il cluster [OC-{Pt6}-CO](CF3SO3)2 si riduce, mantenendo le sue
caratteristiche principali, in due stadi reversibili monoelettronici [43]; anche i
corrispondenti derivati neutri dialogenurici, X-{Pt6}-X (X = Cl, Br, I), si comportano
allo stesso modo [42]. La stabilità delle unità cluster in diversi stati di ossidazione,
inoltre, ha reso più facile la valutazione della comunicazione elettronica lungo una
struttura estesa con misure elettrochimiche relativamente semplici, consentendo
l’ispezione degli effetti dello stato di ossidazione degli atomi di platino sulla
comunicazione elettronica stessa. Misure di questo tipo sono state effettuate su un
sistema modello costituito da una singola unità Pt6 legata attraverso uno spaziatore
etinilico (-C≡C-) ad una o a due sonde redox ferroceniliche [44].
Le potenziali applicazioni di questa tipologia di macromolecole organometalliche,
specialmente su un’eventuale larga scala, sono tuttavia limitate dal costo elevato di un
metallo prezioso come il platino. Da ciò deriva l’importanza nella ricerca di cluster di
metalli di transizione alternativi al platino che possano essere utilizzati come sintoni
per la costruzione di macromolecole con caratteristiche simili o addirittura migliori,
ovvero con possibilità applicative di interesse nell’ambito sia della scienza dei
materiali che in altri settori.

20
1.4 Perché i cluster di ferro
La nascita delle varie generazioni di stelle, dalle prime più ricche di elementi leggeri
come l'idrogeno e di grandi dimensioni fino alle stelle come il sole, accesosi circa 5
miliardi di anni fa, ha arricchito l'universo, tramite vari cicli di fusione, di diversi
elementi chimici: sono nati così i nuclei più pesanti del litio, come il carbonio, l'azoto,
l'ossigeno, il calcio e così via fino al ferro che fra gli elementi pesanti è uno dei più
abbondanti del cosmo, come mostrato in Fig. 1.22: l'ingente presenza di Fe si accorda
con il fatto che esso possiede un nucleo particolarmente stabile [45].
Oltre ad essere uno degli elementi più abbondanti nel cosmo il ferro, appartenente al
gruppo 8 della tavola periodica, è il secondo elemento metallico più abbondante nella
crosta terrestre e viene intensamente estratto (circa 109 t/anno). Queste generiche
informazioni basterebbero per rendere plausibile qualsiasi tentativo di ricerca volto
all’approfondimento della chimica di questo metallo, specialmente nel caso in cui si
vogliano prospettare applicazioni su larga scala. Infatti, l’aspetto dell’abbondanza
sottolineato finora, è legato anche ai costi che, oggi più che mai, hanno un peso non
trascurabile: basti pensare che il platino ha una quotazione che oscilla, ad oggi, intorno
ai 1100 € l’oncia mentre 1 kg di polvere di ferro con una purezza ≥ 99 % costa circa
Figura 1.22 Abbondanza cosmica degli elementi [45].

21
36 €. L’aspetto dei costi unitamente a quello delle caratteristiche elettrochimiche e
magnetiche [46] ha stimolato l’indagine sui cluster di ferro da usare come sintoni
organometallici per la sintesi di composti oligomerici in analogia con quanto fatto, nel
laboratorio in cui è stato svolto il presente lavoro di tesi, con i cluster di platino.
1.4.1 Cluster carbonilici di ferro
In letteratura sono noti molti esempi di cluster di ferro con differente nuclearità [47].
Tra questi, maggiore attenzione è stata data a cluster carbonilici con l’atomo di C
interstiziale come ad esempio il cluster pentanucleare Fe5C(CO)15 a 74 elettroni (Fig.
1.23), sintetizzato per la prima volta da Dahl et al. nel 1962: questi lo isolarono in
maniera inaspettata durante gli studi volti ad indagare le reazioni tra alchini ed
Fe3(CO)12. In particolare, la formazione di tale cluster fu osservata in seguito al
riscaldamento del cluster trinucleare con metilfenilacetilene o 1-pentino in etere di
petrolio e, nei casi più favorevoli, esso si ottenne con una resa dello 0.5 % [48].
Il miglioramento dei metodi di sintesi e, quindi, delle rese [49] ha consentito
un’indagine più profonda della chimica di questo cluster. Ad esempio, Poë et al. hanno
effettuato studi di tipo cinetico che hanno permesso di confrontare la reattività con
l’analogo strutturale Ru5C(CO)15 [50] e di vedere se, come il carbonile trinucleare
Fe3(CO)12, Fe5C(CO)15 mostra scarsa tendenza a dare reazioni con fosfine P-donatrici
Figura 1.23 Struttura di Fe5C(CO)15. Gli atomi di ferro sono localizzati sui vertici di una piramide
a base quadrata: il frammento Fe4C(CO)12 è a simmetria C4v mentre il frammento apicale Fe(CO)3
è a simmetria C3v [48].

22
o con altre specie nucleofile: si è dedotto che tale cluster, come pure l’analogo
pentanucleare di rutenio, risulta particolarmente suscettibile a subire attacchi da parte
di specie nucleofile attraverso processi di tipo associativo [51]. Nell’ambito dei cluster
carbonilici, inoltre, rivestono un ruolo non trascurabile molte specie anioniche come
ad esempio [Fe2(CO)8]2-, [Fe3(CO)11]
2-, gli anioni tetranucleari [Fe4(CO)13]2- e
[Fe4C(CO)12]2-, [Fe5C(CO)14]
2- e l’esanucleare [Fe6C(CO)16]2- [52]. Queste specie
anioniche sono spesso i reagenti di partenza per la sintesi di cluster bimetallici
contenenti, oltre al ferro, altri metalli di transizione come Cr, Mo, W, Rh, Ir, Pd, Ni
[49a], Cu [53, 49a], Pt (Fig. 1.24) [54] e Hg[55].
Relativamente ai cluster bimetallici che è possibile sintetizzare a partire dai cluster
anionici del ferro vanno citati anche quelli Fe-Au. Un esempio è dato dalla reazione
fra [Et4N]2[F5C(CO)14] e (ClAu)2(L-L) (L-L = dppm, dppe, dppp): quando il rapporto
fra i reagenti è 1:1 si ottiene [F5C(CO)14(Au2L-L)], che presenta una struttura costituita
da un core {Fe5C} con geometria di piramide a base quadrata in cui un atomo di Au
cappa la faccia triangolare ai cui vertici stanno tre atomi di Fe mentre l’altro Au cappa
la faccia triangolare data dai due atomi di Fe basali e dall’atomo di Au apicale (Fig.
1.25) [52].
Figura 1.24 Struttura di PtFe4C(CO)12(PMe2Ph)2: tale cluster si ottiene con rese molto basse [54];
ciò limita le indagini sulla reattività.
Figura 1.25 Struttura dello scheletro metallico di [F5C(CO)14(Au2L-L)] [52].

23
Nel caso in cui il rapporto fra i reagenti sopra citati è 2:1 si ottiene, invece,
[Et4N]2[{Fe5C(CO)14Au}2L-L], nel quale due frammenti ottaedrici {Fe5CAu} sono
legati attraverso la difosfina (Fig. 1.26) [52].
Un limite di questa classe di composti citati ma anche di quelli che sono stati oggetti
di studio in questo lavoro di tesi, è costituito dalla sensibilità più o meno marcata
all’aria di questa serie di composti. Ad ogni modo, come mostrato in letteratura, tali
cluster carbonilici del ferro con l’atomo di C interstiziale, che stabilizza il cluster verso
reazioni che potrebbero portare alla sua degradazione [54], risultano adatti per
un’indagine volta ad una migliore comprensione della loro reattività necessaria al fine
di poterne valutare i possibili usi come sintoni per la costruzione di specie polimeriche
con proprietà tutte da studiare. Si vedranno più avanti le strategie sperimentali adottate
e le caratterizzazioni effettuate.
Figura 1.26 Strutture dei due frammenti ottaedrici F5CAu legati attraverso la difosfina [52].

24
2
SCOPO DELLA TESI
In questo lavoro di tesi è stata svolta un’indagine preliminare sulla sintesi e la reattività
di cluster carbonilici di ferro contenenti un atomo di carbonio interstiziale, al fine di
valutare il loro possibile impiego come sintoni per la costruzione di specie
oligomeriche e polimeriche, in analogia con quanto fatto negli ultimi anni con i cluster
di platino nel laboratorio in cui è stato svolto il presente lavoro. Ciò con lo scopo ultimo
di ottenere, a partire dagli eventuali sintoni, sistemi conduttori o semiconduttori di
basso costo e quindi con maggiori potenzialità applicative rispetto ai derivati analoghi
noti in letteratura che impiegano metalli molto più costosi [25, 26, 28, 29, 35, 42]. I
cluster carbonilici scelti contengono un atomo di carbonio interstiziale, che in genere
stabilizza il cluster verso reazioni che potrebbero portare alla sua degradazione [54].
Nel presente lavoro di tesi, è stata anche studiata l’elettrochimica di alcuni dei
composti sintetizzati tramite misure di voltammetria ciclica e spettroelettrochimica.

25
3
RISULTATI E DISCUSSIONE
3.1 Sintesi e reattività dei cluster carbonilici di ferro
3.1.1 Sintesi, caratterizzazione e reattività del cluster Fe5C(CO)15 (1)
Isolato casualmente nel 1962 da Dahl et al. [48], il cluster pentanucleare a 74 elettroni
di valenza Fe5C(CO)15 (1) è stato ottenuto per reazione tra Fe(CO)5 e Na2Fe(CO)4 . 1.5
diossano. Il ferro pentacarbonile è un liquido di colore giallo chiaro, tossico ed
infiammabile come altri metallocarbonili e come lo stesso Na2Fe(CO)4 . 1.5 diossano,
che s’incendia per esposizione all’aria. Il reagente viene molto utilizzato in sintesi
metallorganica come materiale di partenza per la sintesi sia di altri metallocarbonili a
base di ferro che di cluster eterometallici contenenti, oltre al ferro e al CO, metalli e
leganti differenti [56]. A causa delle caratteristiche dei reagenti di partenza, la
soluzione in diglima degli stessi è stata preparata lavorando sotto atmosfera di N2.
Dopo 5 h a 150 °C ed una successiva a 160 °C, in un bagno ad olio in condizioni di
buona agitazione, la soluzione è stata raffreddata fino a T ambiente: l’olio ottenuto per
stratificazione di un forte eccesso di n-esano è stato estratto con acqua deionizzata e
degassata a dare una soluzione di colore viola intenso contenente il sale di sodio
dell’anione a 6 atomi di ferro [Fe6C(CO)16]2-.
Tale anione non è stato isolato ma degradato ossidativamente ad 1, in accordo con
quanto riportato da Tachikawa et al. [49a], usando una soluzione acquosa concentrata
di FeCl3, secondo lo schema di reazione riportato in Fig. 3.1.
Figura 3.1 Schema di sintesi di Fe5C(CO)15 [49a].
1

26
Il cluster pentanucleare di ferro è stato recuperato, dopo work up, con una resa del
71%. La procedura seguita differisce da quella riportata da Poë et al. [51a] che prevede
un ulteriore step di isolamento del sale di ammonio del cluster esanucleare anionico e
la sua successiva degradazione ossidativa al prodotto 1 per reazione con (C7H7)BF4. A
causa della sensibilità all’aria, Fe5C(CO)15 è stato conservato in fiale sigillate in
atmosfera di N2. Tale sensibilità, già nota in letteratura [49a], è stata confermata
esponendo all’aria una porzione di solido 1: già dopo un minuto, il solido nero inizia
ad assumere sfumature arancio scuro che diventano sempre più evidenti col passare
del tempo. Dopo circa dieci minuti tutta la massa di 1 assume una colorazione arancio
scuro e il suo spettro IR non presenta più alcun segnale nella finestra spettrale
analizzata. Per questo motivo, tutte le caratterizzazioni sono state effettuate preparando
i campioni in atmosfera inerte.
Lo spettro IR della soluzione ottenuta sciogliendo in CH2Cl2 una porzione del solido
nero recuperato al termine della procedura sperimentale, riportato in Fig. 3.2, rivela la
presenza delle bande carboniliche del prodotto desiderato (νCO = 2099 (d), 2051 (mf),
2032 (f), 2012 (d) e 1992 (md) cm-1), in accordo con quanto riportato in letteratura
[49a] e l’assenza di altre bande imputabili a sottoprodotti eventualmente presenti nel
solido isolato dopo il work up.
Figura 3.2 Spettro IR a T ambiente di Fe5C(CO)15.

27
Lo spettro 13C NMR di 1 (CH2Cl2, 293 K) mostra due singoletti a 209.1 e 212.8 ppm,
rispettivamente dovuti ai 12 carbonili coordinati, 3 per ciascuno, ai 4 atomi di ferro
basali ed ai 3 carbonili coordinati all’atomo di ferro apicale nella struttura piramidale
a base quadrata di questo cluster (§ 1.4.1, Fig. 1.23). Anche in questo caso i valori
ottenuti sono in accordo con quelli riportati in letteratura [49a] e confermano la natura
del cluster sintetizzato.
Come già anticipato (§ 1.4.1), Fe5C(CO)15 tende a subire attacchi da parte di specie
nucleofile attraverso processi di tipo associativo. Negli studi effettuati da Poë et al.
con nucleofili P-donatori come PPhMe2, PPh3, PPhCy2, PPh(OMe)2 e altri, ovvero con
specie caratterizzate da una diversa capacità σ-donatrice e da un diverso valore
dell’angolo conico (θ) di Tolman, si è dedotto che 1, in presenza di un equivalente di
legante P-donatore (L) caratterizzato da un valore di θ non troppo elevato, dà luogo a
reazioni di sostituzione di un legante carbonilico. Il primo stadio della reazione è
costituito dalla coordinazione della fosfina a dare un addotto a 76 elettroni di valenza
che, nel secondo stadio, perde un legante carbonilico a dare il prodotto monofosfina-
sostituito come di seguito riportato:
L’indagine degli autori sopra citati è di rilievo poiché ha messo in evidenza, per quanto
riguarda la reattività di 1 con fosfine, la relazione che lega il valore di θ del legante L
con le dimensioni del core Fe5C. Confrontando la cinetica di reazione di 1 con quella
del suo analogo di rutenio Ru5C(CO)15 nelle reazioni con gli stessi leganti P-donatori,
gli autori hanno osservato che il cluster di ferro dà generalmente reazioni di un fattore
~103 più lente e ciò è tanto più evidente quanto maggiore è il valore di θ associato alla
fosfina. Questo è stato chiaramente correlato alla struttura di Fe5C(CO)15 che presenta
distanze di legame Fe-Fe significativamente più corte rispetto alle corrispondenti
distanze Ru-Ru del cluster di rutenio: in particolare, la distanza tra gli atomi di Fe
basali è di 23 pm più piccola mentre quella tra il ferro apicale e uno di quelli basali è
(3.1)
(3.2)

28
di 16 pm più piccola rispetto alle distanze Ru-Ru corrispondenti [51a]. Le ridotte
dimensioni del core metallico Fe5C e l’effetto sterico che ne può conseguire
giustificano il fatto che con nucleofili di grandi dimensioni (θ > 136°) le reazioni di
sostituzione sono molto lente e in alcuni casi si può osservare la degradazione dello
stesso cluster prima che abbia luogo la reazione. Di conseguenza è molto difficile, nel
caso del cluster 1, l’isolamento di un prodotto monosostituito contenente ligandi di
grandi dimensioni come le fosfine terziarie PPh3 o PPhCy2. Ciò è stato confermato
anche in questo lavoro di tesi analizzando la reazione fra Fe5C(CO)15 e PPh3: l’analisi
IR effettuata su un campione della soluzione alcune ore dopo aver introdotto in un
pallone 1 equivalente di cluster e di PPh3 in CH2Cl2, non ha infatti mostrato alcun
cambiamento nei segnali di 1, mentre prelievi analizzati nei giorni successivi hanno
mostrato una graduale riduzione fino a scomparsa delle bande di 1, un dato che,
unitamente alla scomparsa dei segnali dovuti ai carbonili nello spettro 13C NMR, ha
fatto presumere la decomposizione del cluster di ferro. La reazione è stata inoltre
provata più volte variando parametri sperimentali come la temperatura e le
concentrazioni dei reagenti di partenza, ma conduce sempre allo stesso esito finale.
3.1.2 Sintesi, caratterizzazione e reattività del cluster Fe5C(CO)14(PHtBu2) (2)
Come già anticipato (§ 3.1.1), il cluster pentanucleare Fe5C(CO)15 può subire attacchi
da parte di nucleofili P-donatori non troppo ingombranti [51a] attraverso processi di
tipo associativo. Mentre, tuttavia, la reattività di 1 con fosfine terziarie è stata indagata
in dettaglio [51], non sono ad oggi noti studi sulla reattività con fosfine secondarie.
L’impiego di fosfine secondarie (o primarie) potrebbe essere un utile mezzo per
accrescere la nuclearità del cluster. Il legame P-H delle fosfine è in effetti un centro
reattivo. Può essere infatti deprotonato con una base forte, per dare un fosfuro
fortemente nucleofilo che può reagire con un altro cluster elettrofilico (Fig. 3.3a).
Inoltre, il debole legame P-H dà facilmente reazioni di addizione ossidativa a centri
metallici elettron-ricchi generando un idruro e un fosfuro che ha una grande preferenza
per la coordinazione a ponte con metalli di fine transizione (Fig. 3.3b).

29
Sia il primo tipo di approccio [57a-e], che il secondo [57f-h] sono già stati impiegati in
letteratura per costruire sistemi a nuclearità più elevata (di ferro o di altri centri
metallici) a partire da sintoni più piccoli. Abbiamo quindi deciso di tentare la sintesi
del cluster monosostituito Fe5C(CO)14(PHtBu2) (2), che è stato in effetti ottenuto per
reazione tra 1 e la fosfina secondaria PHtBu2, dopo aver sciolto in CH2Cl2 i reagenti di
partenza in quantità equimolare (Fig. 3.4).
Il cluster monosostituito 2 è stato isolato dopo 5 h di agitazione della soluzione di
partenza a temperatura ambiente, quando il singoletto dovuto alla fosfina libera, che
cade a 21 ppm, non è stato più osservato nello spettro 31P{1H} NMR del grezzo di
reazione e l’analisi IR non ha più mostrato le bande carboniliche del precursore. Il
composto 2, recuperato con una resa quantitativa, è stato conservato in fiale sigillate
sotto N2. Anche in questo caso, infatti, il composto sintetizzato è sensibile all’aria e
decompone in maniera analoga al cluster precursore.
Lo spettro IR della soluzione di 2 in CH2Cl2 rivela una maggiore complessità delle
bande carboniliche (νCO = 2081 (m), 2053 (m), 2037 (f), 2028 (f), 2013 (f), 2002 (f, br)
e 1982 (m, br) cm-1), in accordo con la diminuzione di simmetria che ha luogo a seguito
della sostituzione di un carbonile con il legante σ-donatore PHtBu2. Oltre a ciò, quello
che si può osservare dallo spettro riportato in Fig. 3.5 è una diminuzione delle
frequenze associate agli stiramenti carbonilici, prevedibile tenendo conto della
sostituzione di un legante π-acido con uno σ-basico.
Figura 3.4 Schema di sintesi di Fe5C(CO)14(PHtBu2).
2
Figura 3.3 Schemi di reazione per specie con una fosfina secondaria coordinata: a) deprotonazione
e successiva reazione con un centro elettrofilico; b) addizione ossidativa del legame P-H.

30
Lo spettro 31P{1H} NMR di 2 (CH2Cl2, 293 K) presenta un singoletto a 76.0 ppm.
Come visibile in Fig. 3.6, nello spettro 31P NMR accoppiato con il protone questo
segnale si presenta come un doppietto (1JP-H = 318 Hz) a causa dell’accoppiamento
dell’atomo di fosforo della fosfina coordinata con il protone adiacente.
La sintesi di questo cluster evidenzia come l’ingombro sterico del legante nucleofilo
giochi un importante ruolo nel decorso della reazione; a differenza di quanto osservato
per la reazione con la fosfina terziaria ingombrata PPh3, che ha un angolo conico molto
Figura 3.6 Spettri 31P{1H} NMR e 31P NMR di Fe5C(CO)14(PHtBu2).
Figura 3.5 Spettro IR a T ambiente di Fe5C(CO)14(PHtBu2).

31
grande (θ = 145°), in questo caso la reazione porta al prodotto finale in condizioni
blande.
Prove di deprotonazione della fosfina coordinata nel cluster 2 sono state effettuate nel
tentativo di ottenere un fosfuro che potesse dare una reazione di sostituzione di un
legante carbonilico su di un’altra unità cluster, raddoppiando la nuclearità del cluster.
Queste prove sono state eseguite facendo reagire 2 con basi forti come NaH o nBuLi,
in differenti condizioni di T, ma in tutte le prove la perdita dei segnali carbonilici
all’IR, e la scomparsa del segnale della fosfina nello spettro 31P NMR ha fatto
presumere la decomposizione del cluster. Riscaldando soluzioni di 2 in assenza di basi
non si sono avute indicazioni che lasciassero supporre il procedere di una addizione
ossidativa, ma anche in questi casi si è osservata la decomposizione del reagente.
3.1.3 Sintesi e caratterizzazione del cluster [Et4N]2[Fe6C(CO)16] (3)
Il cluster esanucleare [Et4N]2[Fe6C(CO)16] (3) è stato sintetizzato usando la stessa
procedura seguita per la preparazione del composto 1 fino all’ottenimento della
soluzione acquosa di colore viola intenso contenente il sale di sodio dell’anione a 6
atomi di ferro [Fe6C(CO)16]2-: questo è stato precipitato dalla soluzione acquosa come
un solido nero/viola con una resa del 79% per aggiunta di un eccesso di Et4NI (Fig.
3.7).
Anche questo cluster è noto in letteratura [49a, 58a] ed è sensibile all’aria, ma in modo
meno marcato rispetto al cluster pentanucleare. Una porzione solida di 3 esposta
all’aria resiste senza subire processi di ossidazione o decomposizione evidenti per
alcune ore, tuttavia, dopo un giorno di esposizione all’aria, il solido nero/viola ha
assunto una consistenza polverosa ed una colorazione arancio che, unitamente alla
scomparsa delle bande carboniliche all’IR e dei segnali dell’anione [Fe6C(CO)16]2- al
13C NMR, ha confermato l’avvenuta decomposizione. La natura dei prodotti di
decomposizione non è stata indagata.
Figura 3.7 Schema di sintesi di [Et4N]2[Fe6C(CO)16].
3

32
La struttura del cluster 3 è nota [58b], lo scheletro di atomi di ferro nell’anione a 86 e-
[Fe6C(CO)16]2- presenta una geometria ottaedrica lievemente distorta con l’atomo di C
carburico, che stabilizza la struttura del cluster [54], leggermente spostato rispetto al
centro dell’ottaedro [58b]; un solo atomo di Fe coordina tre carbonili terminali mentre
gli altri cinque ne coordinano due (Fig. 3.8). Sono, inoltre, presenti tre leganti CO
‘semi-bridging’ (Fig. 3.8).
L’unità asimmetrica contiene poi due cationi tetraetilammonio indipendenti [58b]. Per
quanto riguarda la caratterizzazione spettroscopica, l’analisi dello spettro IR rivela la
presenza di bande carboniliche che, rispetto ai cluster visti in precedenza (§ 3.1.2, §
3.1.1), cadono a frequenze più basse (νCO = 2032, 1967, 1930, 1892, 1767 cm-1) in
virtù della natura anionica del cluster esanucleare.
Lo spettro 13C{1H} NMR di 3 (CH2Cl2, 293 K) mostra, oltre ai segnali relativi agli
atomi di carbonio dei cationi, un singoletto a 228.5 ppm, attribuito agli atomi di
carbonio dei leganti CO coordinati terminalmente, un singoletto a 256.8 ppm
assegnato ai carbonili ‘semi-bridging’ ed infine un segnale di piccola intensità a campi
bassi (δ = 484.0 ppm) associato al carbonio carburico. Il composto 3 è stato utilizzato
come precursore per la sintesi di altri composti (§ 3.2, § 3.3.1 e § 3.3.2).
3.1.4 Sintesi e caratterizzazione del cluster [Et4N]2[Fe5C(CO)14] (5)
Come mostrato nello schema in Fig. 3.9, dalla riduzione di 1 con [Fe(CO)4]2- è stato
ottenuto il cluster anionico a 74 e- [Fe5C(CO)14]2- [49a].
Figura 3.8 Struttura dell’anione [Fe6C(CO)16]2- [58b].
Figura 3.9 Schema di sintesi di [Fe5C(CO)14]2- [49a].

33
La reazione tra Fe5C(CO)15 e Na2Fe(CO)4 . 1.5 diossano in diglima decorre in
condizioni blande, in accordo con quelle descritte da Tachikawa et al. [49a] (§ 5.4.5).
Il composto 5 è stato precipitato in seguito all’aggiunta, goccia a goccia, di una
soluzione acquosa di Et4NCl nella soluzione in diglima ed è stato recuperato come
solido nero/marrone con una resa dell’89%. Anche [Et4N]2[Fe5C(CO)14] è sensibile
all’aria ed ha mostrato un comportamento simile a quello del composto 1.
Lo spettro IR della soluzione di 5 in CH2Cl2 presenta le bande relative agli stiramenti
carbonilici del prodotto desiderato (νCO = 2028, 1965, 1953sh, 1948, 1923sh cm-1), in
accordo con quanto riportato in letteratura [49a]: i segnali risultano spostati a frequenze
più basse rispetto ai segnali dell’analogo cluster neutro 1, in sintonia con la natura
anionica di questo composto.
In accordo con quanto riportato in letteratura [49a], lo spettro 13C{1H} NMR di 5 (THF,
293 K) mostra, oltre ai segnali relativi agli atomi di C dei due cationi, due singoletti:
uno a 223.4 ppm, che integra per tre atomi di carbonio (i tre carbonili coordinati al
ferro apicale), ed uno a 227.2 ppm, che integra per 11 atomi di carbonio (i carbonili
coordinati agli atomi di ferro basali).
Il cluster [Fe5C(CO)14]2- è un buon nucleofilo e viene in genere utilizzato [49a, 54, 55b,
59], come precursore per la sintesi di nuovi cluster ottaedrici contenenti, oltre allo
scheletro pentanucleare di atomi di ferro, altri metalli (come ad esempio Au, Hg, Mo
e Pt) che permettono di aumentare la nuclearità e di espandere la struttura del cluster
precursore. Un limite relativo alla sintesi di sistemi di questo tipo è però costituito
dalle laboriose procedure di recupero dei prodotti finali e soprattutto dalle basse rese
che limitano le possibilità di indagine sulla reattività di questi composti.
3.2 Sintesi di sali contenenti cluster nel catione e nell’anione
Sali organometallici nei quali entrambi gli ioni sono dei cluster sono abbastanza rari
[60]. Tra i primi esempi di questo tipo di sali vanno citati quelli preparati da Green et
al. nel 1987 [61], nei quali sia il catione che l’anione sono costituiti da cluster con
formula generale [M4X4Y4]n+/- (M = Fe, Cr, Mo; X = µ3-S, µ3-Se; Y = η5-C5H4R), e

34
quello riportato da Podlahová et al. nel 1989 [62] in cui il catione è il cluster triangolare
[Rh3(µ3-PMe)(PMe3)4(CO)5]
+ mentre l’anione è il cluster ottaedrico bimetallico
[FeRh5(PMe3)(CO)15]-.
Sali organometallici del tipo descritto sono stati in genere ottenuti come intermedi in
tentativi di sintesi di cluster a nuclearità più elevata [60a, 63], effettuati facendo reagire
un cluster anionico con un cluster cationico. Un esempio di quanto detto è fornito dalla
sintesi del cluster bimetallico [Ru6C(CO)16Pt3(dppm)2], preparato da Koshevoy et al.
[60a] proprio a partire dalla coppia ionica di cluster [Ru6C(CO)16]2-
[Pt2(CO)2(dppm)2]2+, in accordo con lo schema di reazione riportato in Fig. 3.10.
Riguardo ad una coppia ionica di cluster, va detto che mentre in alcuni casi i due ioni
rimangono come entità indipendenti [60c, d, 62], in altri si può avere trasferimento
elettronico fra i due cluster: ciò può comportare una semplice variazione nella struttura
di uno o di entrambi i cluster [60a, b] o rappresentare il primo stadio della reazione che
dai due ioni intermedi porta ai prodotti finali, in genere costituiti da cluster a nuclearità
più alta [60a, e,63].
Anche in questo lavoro di tesi sono state effettuate indagini riguardo alla sintesi di sali
caratterizzati da una coppia ionica di cluster. Sfruttando come precursori i cluster
dianionici del ferro [Fe6C(CO)16]2- e [Fe5C(CO)14]
2-, si è provato ad ottenere questi
sali mediante sostituzione di entrambi i cationi tetraetilammonio, nei composti 3 e 5,
con lo ione [Pt3(µ-PtBu2)3(CO)3]+ (Fig. 1.192). In un primo esperimento i composti 3
e [Pt3(µ-PtBu2)3(CO)3]CF3SO3, preparato in accordo con la procedura riportata in
letteratura [64], sono stati sciolti in pochi mL di acetone sotto atmosfera di N2: sulla
soluzione di colore viola ottenuta è stato quindi stratificato etere dietilico. Dopo 10
giorni, è stata osservata la formazione di cristalli neri a scaglie, i quali sono stati
recuperati ed infialati. L’analisi della struttura mediante diffrazione a raggi X su
Figura 3.10 Schema di sintesi del cluster bimetallico [Ru6C(CO)16Pt3(dppm)2]
[60a].

35
singolo cristallo effettuata dal Prof. S. Zacchini, ha mostrato che è stato ottenuto un
sale misto nel quale uno soltanto dei cationi tetraetilammonio è stato sostituito dal
catione [Pt3(µ-PtBu2)3(CO)3]+. Nota la composizione, è stato possibile valutare che
[Pt3(µ-PtBu2)3(CO)3][Et4N][Fe6C(CO)16] (4) è stato ottenuto con una resa (basata su
3) del 94%.
Lo spettro 31P{1H} NMR di 4 (CH2Cl2, 293 K) presenta un singoletto che cade a 155.4
ppm relativo ai fosfuri a ponte fra gli atomi di platino del catione; come visibile in Fig.
3.11, il segnale presenta i satelliti attesi a causa dell’accoppiamento con il Pt (1JP-Pt =
1652 Hz). I valori ottenuti sono risultati in accordo con quelli riportati in letteratura
[64] per il composto [Pt3(µ-PtBu2)3(CO)3]CF3SO3, suggerendo che non ci sia alcuna
interazione elettronica tra il cluster cationico e il cluster anionico.
La mancanza di interazione tra le due unità cluster è stata confermata dallo spettro IR
della soluzione di 4 in CH2Cl2, che rivela la presenza delle bande associate agli
stiramenti carbonilici dei due cluster senza che vi sia alcuna modifica nei valori di
frequenza rispetto agli spettri IR dei precursori analizzati separatamente. In particolare,
come visibile in Fig. 3.12, a 2063 cm-1 si trova la banda relativa agli stiramenti
carbonilici del catione di platino, in accordo con quanto riportato in letteratura (νCO
[Pt3(µ-PtBu2)3(CO)3]CF3SO3 = 2064 cm-1) [64], mentre gli altri segnali (νCO = 2032,
Figura 3.11 Spettro 31P{1H} NMR di [Pt3(µ-PtBu2)3(CO)3][Et4N][Fe6C(CO)16].

36
1966, 1930sh e 1768 cm-1) sono quelli associati agli stiramenti carbonilici del cluster
di ferro.
Un esperimento analogo a quello che ha portato al sale misto 4 è stato effettuato per la
sintesi di un sale con [Fe5C(CO)14]2- come anione: in questo caso non siamo riusciti ad
ottenere un solido puro e non sono state effettuate ulteriori indagini a riguardo.
3.3 Sintesi di cluster bimetallici
Cluster metallici ad alta nuclearità contenenti centri metallici diversi hanno suscitato
un certo interesse in virtù delle loro possibili differenti applicazioni [65, 49a, 52, 53,
54, 55]. Dal nostro punto di vista, l’interesse nei confronti di questa classe di composti
è legato alla necessità di disporre di specie che possono dare reazioni di sostituzione
di un legante in modo ben definito: nell’ambito dei cluster carbonilici studiati, ciò
diventa possibile funzionalizzando il cluster carbonilico con frammenti elettrofilici M-
L+ [66, 67, 68].
In questo contesto si inserisce l’indagine sulla sintesi e sulla reattività di un cluster
bimetallico ferro-rame caratterizzato dalla presenza di un’unità -CuCl che può
rappresentare un punto di attacco al fine di aumentare la nuclearità del sistema. Allo
scopo di ottenere strutture oligomeriche, Della Pergola et al. hanno, ad esempio,
studiato le reazioni di sostituzione dei leganti cloruro con leganti N-, P-, S- donatori
Figura 3.12 Spettro IR di [Pt3(µ-PtBu2)3(CO)3][Et4N][Fe6C(CO)16].

37
sui cluster [Fe4C(CO)12(CuCl)2]2- e [Fe5C(CO)14(CuCl)]2- [66]. L’indagine ha
mostrato che in molti casi la reazione di sostituzione del legante cloruro è
accompagnata da riarrangiamenti strutturali che comportano la perdita o il guadagno
di atomi di metallo a dare strutture del tipo Fe4Cu/Fe4Cu3. Ciò è quanto accade nella
reazione del cluster [Fe5C(CO)14(CuCl)]2- con pirazina, dove la sostituzione del
legante cloruro è accompagnata dalla perdita di un frammento Fe(CO)2 sostituito da
Cu-N, a dare il composto dimerico riportato in Fig. 3.13 [66].
Anche il mercurio può legarsi a diversi cluster di metalli di transizione attraverso
frammenti Hg(m)+ di varia natura (m = Cl [68b, 72, 73], Br, CN [72], Mo(CO)3Cp,
W(CO)3Cp, Mn(CO)5, Co(CO)4, Fe(CO)2Cp [52, 55]). Rossell et al. [55a] riportano,
ad esempio, la sintesi di una serie di cluster ferro-mercurio di formula generale
[Et4N][Fe6C{Hg(M)}(CO)16] generati dalla reazione dell’anione esanucleare
[Fe6C(CO)16]2- con ClHg(M) (M = Mo(CO)3Cp, W(CO)3Cp, Mn(CO)5). Sfruttando
tale reattività si è provato ad introdurre un frammento elettrofilico –HgCl all’interno
del cluster, in modo da ottenere dei sistemi analoghi ai cluster ferro-rame ottenuti da
Della Pergola et al.
3.3.1 Sintesi, caratterizzazione e reattività di [Et4N]2[Fe5C(CO)14(CuCl)] (6)
L’associazione di ferro e rame in composti eterometallici è rilevante in differenti
campi, tra i quali quello della chimica bioinorganica [69], della catalisi eterogenea
[70a] e della catalisi omogenea [70b]. Anche nell’ambito dei cluster carbonilici, la
combinazione di rame e ferro ha suscitato grande interesse sia per quanto riguarda lo
studio delle strutture dei composti risultanti sia per le indagini teoriche sul legame
metallico Cu-Fe che si viene ad instaurare in questi composti [71].
Figura 3.13 Struttura allo stato solido di [{Fe4C(CO)12Cu}2(µ-pyz)]2- [66].

38
L’attenzione rivolta nei confronti del composto [Et4N]2[Fe5C(CO)14(CuCl)] (6), nel
quale il rame mantiene il suo stato di ossidazione formale +1, è scaturita dalla
possibilità di ottenere diversi altri derivati attraverso reazioni di sostituzione del
legante cloruro. Per quanto riguarda la sintesi, [Et4N]2[Fe5C(CO)14(CuCl)] è stato
ottenuto seguendo la procedura riportata da Della Pergola et al. [53], che prevede la
reazione tra il cluster anionico 3 e CuCl in THF a riflusso per 50 minuti; la soluzione
è stata poi raffreddata a T ambiente ed il solvente è stato rimosso sotto vuoto. Il solido
depositato sul fondo del pallone è stato ridisciolto in metanolo e precipitato per
aggiunta di una soluzione acquosa di Et4NCl. Il precipitato di colore nero è stato
recuperato con una resa del 71% dopo essere stato filtrato, lavato con piccole porzioni
di acqua degassata e seccato alla pompa meccanica. Data la sensibilità all’aria di 6,
tutte le operazioni sono state condotte in atmosfera inerte ed il composto è stato
conservato in fiale sigillate sotto N2. Il dianione [Fe5C(CO)14(CuCl)]2- è un cluster a
86 e-, formalmente derivato dalla sostituzione di un frammento Fe(CO)2 con
l’isoelettronico CuCl. Tale anione è caratterizzato da una struttura ottaedrica di atomi
di metallo con il carburo interstiziale al centro dell’ottaedro (Fig. 3.14), con uno dei
vertici occupato dall’atomo di Cu, e l’unità Fe5C che compone invece una piramide a
base quadrata. Due carbonili ‘semi-bridging’ si trovano coordinati tra l’atomo di Fe(3)
e quelli di Fe(2) e Fe(4), mentre l’atomo di ferro apicale Fe(1) e quello basale Fe(5)
legano tre carbonili terminali ciascuno (Fig. 3.14) [53].
Figura 3.14 Struttura dell’anione [Fe5C(CO)14(CuCl) ]2- [53].

39
Lo spettro IR (νCO = 2035, 1973, 1957, 1920 e 1779 cm-1) risulta in accordo con quello
riportato in letteratura [53], con le bande dovute agli stiramenti carbonilici a valori di
frequenze che testimoniano la natura anionica del cluster.
Prove di sintesi di derivati alchinilici del composto 6 sono state effettuate in diverse
condizioni di temperatura e di concentrazione delle soluzioni dei precursori. Nello
specifico, sia le prove effettuate usando come reagente di partenza il fenilacetilene, da
solo ed in presenza di trietilammina, che quelle effettuate con il fenilacetiluro di litio
generato in situ a bassa T, non hanno portato alla sostituzione con l’alchinile del
legante cloruro coordinato al rame ma, piuttosto, alla decomposizione del cluster,
come evidenziato dalla colorazione arancio assunta dalla soluzione inizialmente nera
e dalla scomparsa, nello spettro infrarosso, delle bande carboniliche del precursore 6.
3.3.2 Sintesi del cluster [Et4N]2[{Fe6C(CO)16}2(Hg)] (7)
Dalla reazione fra [Et4N]2[Fe6C(CO)16] e HgCl2 in quantità equimolare è stato ottenuto
il composto 7 [52, 55a], che in letteratura era stato ottenuto dalla reazione di 3 con
Hg(NO3)2. Come riportato da Rossell et al. [55a], la soluzione iniziale in THF è stata
mantenuta per 3 giorni sotto agitazione: dopo questo periodo di tempo, lo spettro IR
di ulteriori prelievi non ha più mostrato alcun cambiamento rivelando la presenza dei
segnali del composto 7 già noto. Dopo work-up, 7 è stato recuperato come solido
nero/marrone con una resa del 49%. Lo spettro IR di [Et4N]2[{Fe6C(CO)16}2(Hg)]
mostra segnali (νCO = 2038, 2010, 1988, 1980sh, 1964 e 1819 cm-1) in accordo con
quelli riportati in letteratura [55a].

40
3.4 Misure elettrochimiche
Il comportamento elettrochimico dei complessi organometallici dipende dalla
nuclearità del complesso, dalla natura dei centri metallici e dalle caratteristiche dei
leganti [74]. I potenziali di riduzione o di ossidazione di un composto possono essere
determinati mediante l’uso della voltammetria ciclica [75].
Un aspetto importante da sottolineare è quello della correlazione che esiste, in genere,
tra la struttura e il NEV di un cluster [5a]. Tale aspetto, infatti, comporta che i processi
di riduzione o di ossidazione chimica (o elettrochimica) sono spesso accompagnati da
ulteriori processi che possono essere di semplice variazione della struttura
(isomerizzazione), di frammentazione o di condensazione del cluster [5a]. In alcuni
casi, specie se sono presenti leganti che si dispongono saldamente a ponte tra due o
più centri metallici, il cluster sopporta un numero variabile di elettroni mantenendo
inalterata la sua struttura e funzionando così da riserva di elettroni [76]. Un esempio
in campo bioinorganico di questo comportamento è fornito dalle ferrodoxine, proteine
che contengono nel loro sito attivo un cluster costituito da 4 atomi di ferro e 4 atomi
di zolfo disposti ai vertici di un cubo: grazie alla capacità che questi cluster hanno di
funzionare da riserva di elettroni, le ferrodoxine mediano la biochimica di reazioni di
trasferimento elettronico [77].
3.4.1 Misure di voltammetria ciclica
In questo lavoro di tesi sono stati effettuati studi di voltammetria ciclica sui composti
1, 2, 3 e 4: in particolare, le misure sono state eseguite su soluzioni 10-3 M in CH2Cl2
a temperatura ambiente (usando come elettrolita di supporto [NBu4][PF6] 0.2 M). I
potenziali formali ricavati per i processi di riduzione ed ossidazione (V vs SCE) dei
composti 1, 2, 3 e 4 sono riassunti in Tabella 3.1.

41
Composti Processi di riduzione
E0 [a] (ΔEp) [b]
Processi di ossidazione
E0 [a] (ΔEp) [b]
1 -0.19c; -1.71 (100)* +0.02c; +0.35c
2 -0.45c; -1.72 (99)* +0.01c; +0.34c
3 -1.54c; -1.81c +0.16c; +0.47c
4 -1.24c; -1.55c; -1.83c +0.15c; +0.46c
Il voltammogramma della soluzione in CH2Cl2 del cluster pentanucleare di ferro
Fe5C(CO)15 (1) è riportato in Fig. 3.15.
Il profilo voltammetrico ottenuto per il composto 1 risulta essere in accordo con quello
riportato per Fe5C(CO)15 da Gourdon et al. [78]. Esso mostra una prima onda a -0.19
V associata al processo di riduzione bielettronico irreversibile schematizzato in Fig.
3.16 [78], seguito dalla perdita di un legante carbonilico che porta alla formazione di
[Fe5C(CO)14]2-, specie alla quale sono da attribuire sia la seconda onda di riduzione
reversibile a -1.71 V (ΔEp = 100 mV*), sia le onde di ossidazione irreversibile a +0.02
V e +0.35 V.
Figura 3.15 Voltammogramma di 1 registrato a 0.1 Vs-1 su elettrodo di platino.
[a, b] Misurato a 0.1 Vs-1. [a] Misurato in V, vs SCE. [b] Misurato in mV. c Valori di potenziale per processi
irreversibili. * Sono considerati reversibili picchi con un ΔEp ≤ 120 mV.
Tabella 3.1 Potenziali formali (V vs SCE) dei composti 1, 2, 3, e 4.

42
In Fig. 3.17 si riporta il voltammogramma del cluster Fe5C(CO)14(PHtBu2).
Il voltammogramma del composto 2 si rivela analogo a quello del composto 1: la prima
onda di riduzione a -0.45 V, si trova ad un valore di potenziale più negativo rispetto a
quella del composto 1 in accordo con la presenza di un legante più basico come la
fosfina, rispetto al carbonile π-acido del composto 1. In analogia con 1, questa onda
può essere associata al processo di riduzione bielettronico irreversibile seguito dalla
dissociazione della fosfina coordinata (Fig. 3.18), come sarà successivamente
evidenziato dagli esperimenti di spettroelettrochimica IR (§ 3.4.2), con formazione
dell’anione [Fe5C(CO)14]2- per il quale sono osservati un processo di riduzione e due
di ossidazione a valori di potenziale analoghi a quelli del composto 1, come riportato
in Tabella 3.1.
Figura 3.16 Riduzione bielettronica di Fe5C(CO)15 a E0 = -0.19 V [78].
Figura 3.17 Voltammogramma di 2 registrato a 0.1 Vs-1 su elettrodo di platino.
Figura 3.18 Riduzione bielettronica di Fe5C(CO)14(PHtBu2) a E0 = -0.45 V.
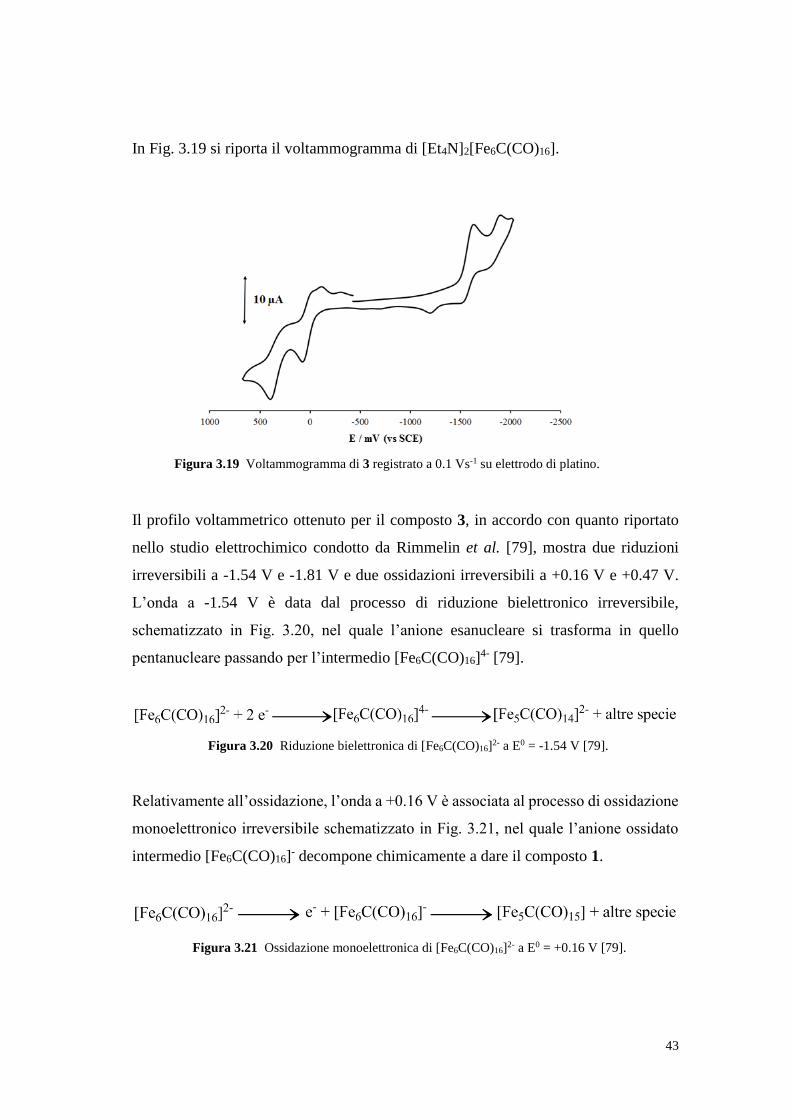
43
In Fig. 3.19 si riporta il voltammogramma di [Et4N]2[Fe6C(CO)16].
Il profilo voltammetrico ottenuto per il composto 3, in accordo con quanto riportato
nello studio elettrochimico condotto da Rimmelin et al. [79], mostra due riduzioni
irreversibili a -1.54 V e -1.81 V e due ossidazioni irreversibili a +0.16 V e +0.47 V.
L’onda a -1.54 V è data dal processo di riduzione bielettronico irreversibile,
schematizzato in Fig. 3.20, nel quale l’anione esanucleare si trasforma in quello
pentanucleare passando per l’intermedio [Fe6C(CO)16]4- [79].
Relativamente all’ossidazione, l’onda a +0.16 V è associata al processo di ossidazione
monoelettronico irreversibile schematizzato in Fig. 3.21, nel quale l’anione ossidato
intermedio [Fe6C(CO)16]- decompone chimicamente a dare il composto 1.
Figura 3.19 Voltammogramma di 3 registrato a 0.1 Vs-1 su elettrodo di platino.
Figura 3.20 Riduzione bielettronica di [Fe6C(CO)16]2- a E0 = -1.54 V [79].
Figura 3.21 Ossidazione monoelettronica di [Fe6C(CO)16]2- a E0 = +0.16 V [79].

44
Anche la seconda ossidazione a +0.47 V è associata ad un processo monoelettronico
che porta nuovamente al cluster pentanucleare [Fe5C(CO)15] (Fig. 3.22) [79].
Infine, le ulteriori onde di minore intensità che si possono osservare nel
voltammogramma di Fig. 3.19 sono associate a processi dati dalle specie ignote che si
generano in soluzione nel corso della riduzione e dell’ossidazione del cluster di
partenza [79].
Il voltammogramma in Fig. 3.23 è quello del sale misto [Pt3(µ-
PtBu2)3(CO)3][Et4N][Fe6C(CO)16].
Nel profilo voltammetrico del sale misto 4 sopra mostrato, si possono osservare tutte
le onde associate ai processi di riduzione ed ossidazione irreversibili dei due cluster
che compongono il sale nell’intervallo di potenziale analizzato. Ad eccezione
dell’onda a -1.24 V, generata dalla riduzione irreversibile trielettronica del cluster
cationico [Pt3(µ-PtBu2)3(CO)3]+ [80], le altre onde presenti nel voltammogramma
Figura 3.22 Ossidazione monoelettronica di [Fe6C(CO)16]2- a E0 = +0.47 V [79].
Figura 3.23 Voltammogramma di 4 registrato a 0.1 Vs-1 su elettrodo di platino.

45
cadono ai valori di potenziale del cluster esanucleare di ferro 3 analizzato prima.
L’assenza di ulteriori picchi dimostra che non vi è alcuno scambio elettronico fra le
due unità cluster che costituiscono il sale.
3.4.2 Esperimenti di spettroelettrochimica
La spettroscopia IR è stata una delle prime tecniche spettroscopiche ad essere associata
all’elettrochimica. La posizione nello spettro delle bande caratteristiche di molti
leganti, come ad esempio quello carbonilico, è infatti sensibile alle variazioni dello
stato d’ossidazione del metallo che si possono avere nel corso di un esperimento
elettrochimico. Grazie al tempo di risposta relativamente breve (~10-13 s) e dello stesso
ordine di grandezza delle velocità dei processi di trasferimento elettronico, tale tecnica
risulta uno strumento importante nella comprensione della natura dei prodotti generati
durante i processi elettrochimici [81]. Gli esperimenti di spettroelettrochimica sono
stati eseguiti su soluzioni in CH2Cl2 dei campioni poste all’interno dell’apposita cella
OTTLE (Optically Trasparent Thin-Layer Cell), registrando dapprima uno spettro del
precursore in modo da monitorare meglio la variazione delle bande caratteristiche del
composto analizzato.
Gli esperimenti di spettroelettrochimica IR sono stati effettuati sui composti 2 e 3. In
Fig. 3.24 si riportano gli spettri IR, registrati al variare del potenziale applicato, del
cluster Fe5C(CO)14(PHtBu2).
Figura 3.24 Variazioni nello spettro IR di Fe5C(CO)14(PHtBu2) registrato con una cella OTTLE
nell’intervallo di potenziale 0.00 V — -0.40 V.

46
Lo spostamento verso potenziali negativi e sempre più prossimi al valore di -0.45 V
(ricavato dall’esperimento di voltammetria ciclica) provoca la riduzione bielettronica
del cluster 2 seguita dalla veloce dissociazione della fosfina coordinata (Fig. 3.18) con
formazione del cluster anionico pentanucleare [Fe5C(CO)14]2-: dallo spettro si osserva,
infatti, una diminuzione nelle intensità delle bande carboniliche del cluster 2 (νCO =
2081, 2053, 2037, 2028, 2013, 2002 e 1982 cm-1), fino quasi alla loro scomparsa ed
un aumento di quelle associate al cluster anionico (νCO = 1965, 1952sh e 1948 cm-1)
[49a]. Quando il verso della scansione viene invertito e si riporta il potenziale a valori
positivi, la reazione torna completamente indietro riformando il cluster di partenza
Fe5C(CO)14(PHtBu2), come risulta visibile dagli spettri in Fig. 3.25, in particolare da
quelli registrati dopo alcuni minuti al potenziale applicato di +0.30 V.
I risultati spettroelettrochimici dimostrano che è possibile invertire il senso della
reazione variando il potenziale applicato in assenza di processi che allontanano la
fosfina secondaria dall’ambiente di reazione. Tale reversibilità è stata dimostrata
anche mediante la riduzione e la successiva riossidazione chimica del cluster 2 (§ 3.5).
Spettri IR nell’intervallo di potenziale applicato 0.00 V — +0.45 V sono stati registrati
per il cluster esanucleare di ferro [Fe6C(CO)16]2- (Fig. 3.26).
Figura 3.25 Variazioni nello spettro IR di Fe5C(CO)14(PHtBu2) registrato con una cella OTTLE
nell’intervallo di potenziale -0.40 V — +0.30 V.

47
Lo spostamento verso valori di potenziale positivi, come evidenziato nel corso della
voltammetria ciclica (Fig. 3.19) e come noto in letteratura [79], comporta l’ossidazione
di [Fe6C(CO)16]2- al cluster pentanucleare [Fe5C(CO)15] (Fig. 3.21 e 3.22). Dagli
spettri registrati in Fig. 3.26 si osserva che, all’aumentare del potenziale applicato alla
cella, si ha una diminuzione sempre più marcata delle bande carboniliche del cluster
esanucleare (νCO = 2032, 1967, 1930sh, 1892, 1767 cm-1) ed un aumento di quelle
associate agli stiramenti carbonilici del cluster pentanucleare neutro 1 (νCO = 2099,
2051, 2032, 2012 cm-1). La presenza di un punto isosbestico segnala la
decomposizione veloce del prodotto di ossidazione del cluster esanucleare senza che
sia possibile rilevarne la presenza.
La scansione inversa nella direzione del potenziale iniziale a 0.00 V ed a potenziali
poco negativi non comporta alcuna variazione nell’ultimo spettro registrato a +0.45 V
e di conseguenza si può concludere che tale ossidazione, come già noto, è irreversibile.
Figura 3.26 Variazioni nello spettro IR del composto 3 registrato con una cella OTTLE
nell’intervallo di potenziale 0.00 V — +0.45 V.

48
3.5 Riduzione chimica e riossidazione di Fe5C(CO)14(PHtBu2)
Al fine di confermare chimicamente quanto osservato nell’esperimento di
spettroelettrochimica su Fe5C(CO)14(PHtBu2), è stata effettuata una prova relativa alla
riduzione chimica ed alla successiva riossidazione di questo cluster. Il processo è stato
seguito attraverso 31P{1H} NMR.
Sapendo che Fe5C(CO)14(PHtBu2) a E0 = -0.45 V subisce una riduzione bielettronica,
in una soluzione di 2 in CH2Cl2 è stata aggiunta una quantità di cobaltocene (riducente
monoelettronico con E0 vs SCE = -0.93 V) doppia in mmol rispetto a quella del cluster
di partenza in modo da ridurlo completamente. Dopo alcuni minuti di agitazione, un
prelievo della soluzione è stato analizzato mediante 31P{1H} NMR (Fig. 3.27).
Come visibile in Fig. 3.27, lo spettro 31P{1H} NMR presenta un singoletto a 20.9 ppm
che è quello del fosforo della PHtBu2 libera e non coordinata. Il segnale del fosforo
della fosfina coordinata nel composto 2 è assente. Si può concludere che tutto il cluster
inizialmente presente è stato ridotto.
Figura 3.27 Spettro 31P{1H} NMR della soluzione di Fe5C(CO)14(PHtBu2) dopo aggiunta di CoCp2.

49
A questo punto, al fine di valutare la reversibilità di questa riduzione, all’interno della
stessa soluzione è stato aggiunto l’ossidante monoelettronico ferricinio (E0 vs SCE =
+0.39 V). Dopo aver aggiunto una quantità equimolare di FeCp2+ rispetto a quella del
cluster iniziale, è stato registrato lo spettro 31P{1H} NMR riportato in Fig. 3.28.
Nello spettro è possibile osservare sia il singoletto della fosfina libera a 20.9 ppm, sia
quello della fosfina coordinata in Fe5C(CO)14(PHtBu2) a 76.0 ppm: ciò dimostra che
l’ossidazione bielettronica del cluster anionico [Fe5C(CO)14]2-, i cui segnali IR erano
stati osservati nel corso dell’esperimento spettroelettrochimico di riduzione di 2,
riforma in presenza di fosfina il composto iniziale. Infatti, dopo aver aggiunto alla
soluzione in CH2Cl2 una quantità complessiva di FeCp2+ pari al doppio in mmol di
quella del cluster di partenza 2, lo spettro 31P{1H} NMR mostra solo il singoletto a
76.0 ppm del fosforo di Fe5C(CO)14(PHtBu2).
Figura 3.28 Spettro 31P{1H} NMR della soluzione di Fe5C(CO)14(PHtBu2) dopo aggiunta di una
quantità equimolare di FeCp2+ rispetto a 2.

50
4
CONCLUSIONI
Il programma di ricerca iniziato con questo lavoro di tesi è finalizzato alla sintesi ed
allo studio della reattività di cluster carbonilici di ferro allo scopo di ottenere possibili
sintoni da usare nella costruzione di strutture macromolecolari. In questo contesto, di
particolare rilievo è la realizzazione di sistemi a basso costo nei quali sia possibile il
trasferimento elettronico fra unità cluster adiacenti. Proprio per questo motivo, le
indagini sono state effettuate sui cluster di un metallo non molto costoso come il ferro
ed in particolare su quelli con un atomo di carbonio interstiziale data la loro maggiore
stabilità strutturale.
Sfruttando gli studi presenti in letteratura [49a, 58a] sono stati sintetizzati due cluster
precursori, [Fe5C(CO)15] (1) e [Fe6C(CO)16]2- (3), che sono stati poi utilizzati come
reagenti di partenza sia per la sintesi dei nuovi composti 2 e 4 sia per quelli già noti in
letteratura 5, 6 e 7 [49a, 53, 55a].
Il cluster Fe5C(CO)14(PHtBu2) (2), ottenuto dalla reazione di 1 con la fosfina
secondaria PHtBu2, è stato sottoposto ad indagini di tipo elettrochimico che hanno
messo in evidenza la possibilità di dissociare reversibilmente la fosfina coordinata
all’unità {Fe5C(CO)14} al variare del potenziale applicato. Tale reversibilità è stata
altresì confermata anche mediante riduzione ed ossidazione chimica. Il cluster 2 è stato
sottoposto anche a prove di deprotonazione della fosfina coordinata nel tentativo di
ottenere un fosfuro che, attraverso una reazione di sostituzione di un legante
carbonilico su di un’altra unità cluster, potesse portare alla formazione di un cluster
dimerico. Queste prove hanno però portato alla decomposizione del cluster evidenziata
dalla perdita dei segnali carbonilici all’IR e dalla scomparsa del segnale della fosfina
nello spettro 31P NMR.
Il sale misto [Pt3(µ-PtBu2)3(CO)3][Et4N][Fe6C(CO)16] (4), preparato a partire da 3 e
[Pt3(µ-PtBu2)3(CO)3]CF3SO3, è stato sottoposto ad indagini voltammetriche che

51
hanno messo in evidenza l’assenza di scambio elettronico fra le due unità cluster che
costituiscono il sale.
Le prove di sintesi di derivati alchinilici mediante sostituzione del legante cloruro nel
cluster [Et4N]2[Fe5C(CO)14(CuCl)], non hanno portato al prodotto atteso con un
legante alchinilico coordinato al rame ma, piuttosto, alla decomposizione del cluster.
Le indagini effettuate sui cluster carbonilici di ferro riportati in questo lavoro
rappresentano comunque un ulteriore passo nella conoscenza della chimica di questi
composti poco studiati. Le difficoltà associate allo studio di questi sistemi, alcuni dei
quali particolarmente sensibili all’aria, e le indagini elettrochimiche hanno evidenziato
i limiti e i pregi di alcuni cluster nella prospettiva di un eventuale loro uso come sintoni
per la costruzione di specie oligomeriche e polimeriche.
Questo studio, in alcuni aspetti pionieristico, rappresenta pertanto un punto di partenza
importante per quanto concerne la conoscenza della reattività e dell’elettrochimica di
alcuni cluster carbonilici di ferro soprattutto per quelli con un legante fosfinico
secondario. Ad ogni modo, trattandosi di un nuovo programma di ricerca sembrano
necessari e indispensabili ulteriori studi al fine di poter esprimere un giudizio rigoroso
sulle reali potenzialità di questi sistemi.

52
5
PARTE SPERIMENTALE
5.1 Generalità
Tutte le operazioni, se non diversamente specificato, sono state condotte in atmosfera
di azoto anidro. La vetreria prima dell’uso è stata tenuta in stufa a 115 °C per circa 1
ora, successivamente evacuata alla pompa meccanica e infine riempita di azoto.
5.2 Solventi e reagenti
I solventi utilizzati, se non diversamente specificato, sono stati anidrificati secondo i
comuni metodi riportati in letteratura, distillati e conservati in atmosfera inerte. In
alcuni casi, sono stati disareati ed usati senza ulteriori purificazioni, mantenendoli su
setacci molecolari in atmosfera inerte. Anche i solventi deuterati sono stati disareati e
conservati in atmosfera inerte su setacci molecolari.
Di seguito si riportano quelli utilizzati e le purificazioni a cui ciascun solvente è stato
sottoposto prima dell’uso.
Diglima, ≥99.5% (Sigma-Aldrich): disareata e conservata su setacci
molecolari in atmosfera inerte.
n-Esano (Sigma-Aldrich): distillato dopo 12 h di riflusso su lega Na/K.
Diclorometano (Sigma-Aldrich): distillato dopo 12 h di riflusso su P2O5.
Toluene (Sigma-Aldrich): distillato dopo 12 h di riflusso su Na.
Acetone (Sigma-Aldrich): distillato dopo 12 h di riflusso su Na2CO3.
Tetraidrofurano (Sigma-Aldrich): distillato su LiAlH4 dopo 12 h di riflusso su
lega Na/K, poi ridistillato dopo altre 12 h di riflusso.
Metanolo, ≥99.9% (Sigma-Aldrich): disareato e conservato su setacci
molecolari in atmosfera inerte.

53
Etere dietilico (Sigma-Aldrich): distillato su LiAlH4 dopo 12 h di riflusso su
lega Na/K, quindi ridistillato dopo altre 12 h di riflusso.
I seguenti reattivi commerciali sono stati impiegati senza ulteriore purificazione:
Fe(CO)5 (Merck), Na2Fe(CO)4 . 1.5 C4H8O2 (Sigma-Aldrich), FeCl3 (Sigma-Aldrich),
(C2H5)4N(I) (Fluka), (C2H5)4N(Cl) (Fluka), CuCl (Aldrich), (C2H5)3N (Sigma-
Aldrich), C6H5CCH (Aldrich), HgCl2 (Sigma-Aldrich).
I seguenti reagenti sono stati preparati in accordo con quanto riportato in letteratura:
[Pt3(µ-PtBu2)3(CO)3]CF3SO3 [64], PHtBu2 [82].
5.3 Misure chimico-fisiche
Gli spettri infrarossi sono stati eseguiti con uno spettrofotometro Perkin-Elmer FT-IR
1725X e con uno spettrofotometro Perkin-Elmer equipaggiato con un accessorio
UATR per campioni solidi. Le abbreviazioni utilizzate nella descrizione degli spettri
sono: mf = molto forte, f = forte, m = medio, d = debole, md = molto debole, sh =
spalla, br = largo.
Gli spettri di risonanza magnetica nucleare sono stati eseguiti con uno spettrometro
Varian Gemini 200 BB e in alcuni casi anche con un Bruker Avance DXR (400 MHz).
Le frequenze (in ppm) sono riferite a (CH3)4Si per 1H e 13C, ed a H3PO4 all’85% per i
nuclei 31P. La simbologia adottata nell’interpretazione degli spettri è la seguente: s =
singoletto, d = doppietto, t = tripletto, dd = doppio doppietto, m = multipletto.
5.3.1 Misure elettrochimiche
Le misure di voltammetria ciclica sono state eseguite con un Princeton Applied
Research (PAR) 273A Potentiostat/Galvanostat, interfacciato con un personal
computer che impiega il PAR M270 Electrochemical Software. Tutte le misure sono
state effettuate utilizzando una cella a tre elettrodi: un disco ed una spirale di platino,
saldati in un tubo di vetro, sono stati usati, rispettivamente, come elettrodo di lavoro e
come controelettrodo. Come elettrodo di riferimento è stato usato un elettrodo di
platino. La cella è stata anidrificata per prolungato riscaldamento in stufa seguito da
raffreddamento sotto vuoto e successivo riempimento con azoto. Le misure sono state
condotte sciogliendo i composti in una soluzione 0.2 M di [Bu4N][PF6] in CH2Cl2,

54
sotto azoto. Prima delle misure, si è provveduto ad una taratura dello strumento
eseguendo dei cicli di voltammetria tra i limiti di interesse anodico e catodico, fino a
quando non si sono più osservati cambiamenti nella corrente di carica dello strumento.
A questo punto, è stato introdotto il substrato ed è stato registrato il voltammogramma
con una velocità di scansione di 100 mVs-1. Dopo l’acquisizione di un numero di
voltammogrammi sufficienti alla caratterizzazione del composto sotto esame, anche a
velocità di scansione differenti, è stata aggiunta una piccola quantità di ferrocene ed è
stato registrato un ulteriore voltammogramma. I valori dei potenziali dei composti
sono stati determinati rispetto alla coppia redox ferrocene/ferrocinio e sono stati riferiti
all’elettrodo a calomelano saturo (SCE) sapendo che nella cella utilizzata la coppia
ferrocene/ferrocinio (Eredox = (Epc + Epa)/2) cade a + 0.390 V vs SCE.
Le misure di spettroelettrochimica registrate all’IR sono state eseguite per mezzo di
una cella OTTLE (Optically Trasparent Thin-Layer Electrode) equipaggiata con
finestre di CaF2, in cui l’elettrodo di lavoro e quello ausiliario sono costituiti da una
piccola griglia di Pt, mentre l’elettrodo di pseudo-riferimento è costituito da un filo di
argento.

55
5.4 Procedure sperimentali
5.4.1 Sintesi di Fe5C(CO)15 (1) [49a]
In un pallone da 250 mL, in atmosfera di N2, sono stati introdotti 15 mL di diglima e
0.979 g (2.829 mmol) di Na2Fe(CO)4 . 1.5 diossano. Alla soluzione di color rosso scuro
ottenuta, dopo breve agitazione, sono stati aggiunti, con l’uso di una siringa attraverso
un setto di gomma posto sul pallone, 1.86 mL (14.145 mmol, ρ = 1.49 g/mL) di
Fe(CO)5. Dopo alcuni minuti di agitazione la soluzione, posta in un bagno ad olio, è
stata scaldata a 150 °C per 5 h e successivamente a 160 °C per 1 h. La soluzione è stata
poi lasciata raffreddare fino a T ambiente sotto agitazione. L’aggiunta di un forte
eccesso di n-esano (100 mL) ha fatto decantare un olio di colore viola scuro che, dopo
aver eliminato il surnatante, è stato lavato più volte con porzioni costituite da alcuni
mL di esano. L’esano residuo è stato poi eliminato sotto vuoto. L’olio è stato, quindi,
estratto con circa 300 mL di H2O deionizzata e degassata suddivisa in tre porzioni: la
soluzione acquosa di colore viola ottenuta è stata raccolta in un pallone da 500 mL. Su
quest’ultima, è stata fatta lentamente gocciolare una soluzione acquosa concentrata di
FeCl3 sotto vigorosa agitazione, con conseguente formazione di un precipitato nero e
sviluppo di CO (visibile attraverso la valvola ad olio). La soluzione col precipitato
nero in sospensione è stata mantenuta ancora sotto vigorosa agitazione per 1 h. A
questo punto, dopo aver filtrato sotto azoto e aver scartato il filtrato, il solido nero sul
filtro è stato lavato con porzioni da 5 mL ciascuna di acqua degassata e lasciato
asciugare sotto vuoto per 1 giorno. Il solido è stato quindi ricristallizzato da toluene
caldo (60 °C) e i cristalli neri ottenuti sono stati infialati, pesati e conservati sotto N2.
Resa (basata su Fe(CO)5): 1.432 g (2.013 mmol), 71%.
13C NMR (CH2Cl2 con capillare di C6D6, 293 K): δ (ppm) = 209.1 (s, 12 CO), 212.8
(s, 3 CO).
IR (cm-1): ν (CO) in CH2Cl2 = 2099 (d), 2051 (mf), 2032 (f), 2012 (d), 1992 (md).
I dati IR ed NMR registrati sono risultati in accordo con i valori riportati in letteratura.

56
5.4.2 Sintesi di Fe5C(CO)14(PHtBu2) (2)
In un pallone da 100 mL sono stati introdotti 30 mL di CH2Cl2, 0.5 g (0.703 mmol) di
Fe5C(CO)15 e 0.13 mL (0.703 mmol, ρ = 0.79 g/mL) di PHtBu2. La soluzione ottenuta
è stata mantenuta sotto agitazione per 5 h a T ambiente: l’avanzamento della reazione
è stato monitorato effettuando ogni ora dei prelievi analizzati mediante 31P{1H} NMR
e IR. Il solido 2 di colore nero, ottenuto per concentrazione a pressione ridotta, è stato
recuperato con una resa quantitativa e conservato in fiale sigillate sotto N2.
31P{1H} NMR (CH2Cl2 con capillare di C6D6, 293 K): δ (ppm) = 76.0 ppm (s, P).
31P NMR (CH2Cl2 con capillare di C6D6, 293 K): δ (ppm) = 76.0 ppm (d, 1JP-H = 318
Hz, PH).
IR (cm-1): ν(CO) in CH2Cl2 = 2081 (m), 2053 (m), 2037 (f), 2028 (f), 2013 (f), 2002
(f, br), 1982 (m, br).
5.4.3 Sintesi di [Et4N]2[Fe6C(CO)16] (3) [49a, 58a]
In un pallone da 250 mL, sotto atmosfera inerte, sono stati introdotti 20 mL di diglima
e 1.123 g (3.245 mmol) di Na2Fe(CO)4 . 1.5 diossano. Alla soluzione di color rosso
scuro ottenuta, dopo breve agitazione, sono stati aggiunti, con l’uso di una siringa
attraverso un setto di gomma posto sul pallone, 2.13 mL (16.225 mmol, ρ = 1.49 g/mL)
di Fe(CO)5. Dopo alcuni minuti di agitazione la soluzione, posta in un bagno ad olio,
è stata scaldata a 150 °C per 5 h e successivamente a 160 °C per 1 h. La soluzione è
stata poi lasciata raffreddare fino a T ambiente sotto agitazione. L’aggiunta di un forte
eccesso di n-esano (100 mL) ha fatto decantare un olio di colore viola scuro che, dopo
aver eliminato il surnatante, è stato lavato più volte con piccole porzioni di esano;
l’esano residuo è stato poi eliminato sotto vuoto. L’olio è stato, quindi, estratto con
circa 300 mL di H2O deionizzata e degassata suddivisa in tre porzioni. Alla soluzione
acquosa di colore viola ottenuta e raccolta in un pallone da 500 mL è stato aggiunto,
sotto agitazione, un eccesso di Et4NI: si è avuta, in tal modo, la precipitazione di un
solido nero/viola che è stato filtrato, lavato con piccole aliquote di acqua degassata e
seccato alla pompa meccanica per 1 giorno. Sono stati ottenuti 2.267 g (2.147 mmol)
di 3, con un resa (basata su Fe(CO)5) del 79%.
13C{1H} NMR (CH2Cl2 con capillare di C6D6, 293 K): δ (ppm) = 6.9 (s, 8 CH3), 51.5
(s, 8 CH2), 228.5 (s, 13 CO), 256.8 (s, 3 µ-CO), 484.0 (s, carburo).

57
IR (cm-1): ν (CO) in CH2Cl2 = 2032 (d), 1967 (f), 1930 (sh), 1892 (d), 1767 (d, br).
I dati IR ed NMR registrati sono risultati in accordo con i valori riportati in letteratura.
5.4.4 Preparazione del sale misto [Pt3(µ-PtBu2)3(CO)3][Et4N][Fe6C(CO)16] (4)
In una provetta, sotto flusso di azoto, sono stati disciolti 0.040 g (0.032 mmol) di
[Pt3(µ-PtBu2)3(CO)3]CF3SO3 [1] e 0.017 g (0.016 mmol) di [Et4N]2[Fe6C(CO)16] in
1.5 mL di acetone. La stratificazione di etere dietilico sulla soluzione viola ottenuta ha
portato, dopo 10 giorni, alla precipitazione di cristalli neri del sale (4). I cristalli sono
stati a questo punto recuperati, infialati sotto N2 e pesati: 0.0305 g (0.015 mmol) con
una resa del 94%.
31P{1H} NMR (CH2Cl2 con capillare di C6D6, 293 K): δ (ppm) = 155.4 (s, P) (1JP-Pt =
1652 Hz).
IR (cm-1): ν (CO) in CH2Cl2 = 2063 (f), 2032 (d), 1966 (f), 1768 (d, br).
5.4.5 Sintesi di [Et4N]2[Fe5C(CO)14] (5) [49a]
In un pallone da 50 mL sono stati introdotti 10 mL di diglima, 0.500 g (0.703 mmol)
di Fe5C(CO)15 e 0.243 g (0.703 mmol) di Na2Fe(CO)4 . 1.5 diossano. Sulla soluzione
nero/rossa ottenuta, dopo 1 h di agitazione, è stata fatta lentamente gocciolare una
soluzione acquosa di Et4NCl, preparata sciogliendo 0.232 g di cloruro di
tetraetilammonio in 10 mL di H2O deionizzata e degassata. Il precipitato di colore
nero/marrone ottenuto è stato filtrato, lavato con etere dietilico fino ad ottenere lavaggi
incolori ed infine seccato alla pompa meccanica. Sono stati ottenuti 0.588 g di (5)
(0.623 mmol) con una resa dell’89%.
13C{1H} NMR (THF con capillare di C6D6, 293 K): δ (ppm) = = 7.0 (s, 8 CH3), 51.8
(s, 8 CH2), 223.4 (s, 3 CO), 227.2 (s, 11 CO).
IR (cm-1): ν (CO) in CH2Cl2 = 2028 (md), 1965 (f), 1952 (sh), 1948 (d), 1923 (sh).
I dati IR ed NMR registrati sono risultati in accordo con i valori riportati in letteratura.
5.4.6 Sintesi di [Et4N]2[Fe5C(CO)14(CuCl)] (6) [53]
In un pallone da 100 mL, munito di refrigerante, sono stati aggiunti 37 mL di THF,
0.367 g (0.348 mmol) di [Et4N]2[Fe6C(CO)16] e 0.054 g (0.545 mmol) di CuCl. La
soluzione ottenuta è stata portata e mantenuta alla temperatura di riflusso per 50

58
minuti, fino a completa conversione. Dopo aver raffreddato a T ambiente, il solvente
è stato rimosso sotto vuoto ed il solido nel pallone ridisciolto in metanolo. A questo
punto, il solido (6) è stato precipitato aggiungendo goccia a goccia una soluzione
acquosa di Et4NCl (1 g in 25 mL): il precipitato nero è stato poi filtrato, lavato con
piccole porzioni di acqua degassata e seccato alla pompa meccanica. Sono stati
recuperati 0.257 g (0.246 mmol) di (6) con una resa del 71%.
IR (cm-1): ν (CO) in THF = 2035 (d), 1973 (mf), 1957 (mf), 1920 (d), 1779 (d, br).
I dati IR registrati sono risultati in accordo con i valori riportati in letteratura.
5.4.7 Sintesi di [Et4N]2[{Fe6C(CO)16}2(Hg)] (7)
In un pallone da 100 mL sono stati introdotti 30 mL di THF, 0.241 g (0.228 mmol) di
[Et4N]2[Fe6C(CO)16] e 0.062 g (0.228 mmol) di HgCl2. La soluzione nero/viola
ottenuta è stata mantenuta sotto agitazione per 3 giorni monitorando la formazione del
prodotto attraverso l’IR. La soluzione è stata successivamente filtrata su Celite ed il
filtrato è stato evaporato a secco. Il solido residuo è stato quindi estratto con 10 mL di
metanolo. La soluzione ottenuta è stata poi ridotta a metà del suo volume iniziale e
conservata in freezer per una notte. Il solido nero/marrone precipitato è stato filtrato e
infine seccato alla pompa meccanica. Sono stati recuperati 0.230 g (0.112 mmol) di
(8) con una resa del 49%.
IR (cm-1): ν (CO) in THF = 2038 (m), 2010 (mf), 1988 (m), 1980 (sh), 1964 (m), 1819
(d).
I dati IR registrati sono risultati in accordo con i valori riportati in letteratura [55a].
5.4.8 Riduzione di Fe5C(CO)14(PHtBu2) (2) e successiva riossidazione
Ad una soluzione ottenuta sciogliendo, all’interno di uno Schlenk, 0.035 g (0.042
mmol) di Fe5C(CO)14(PHtBu2) in 2 mL di CH2Cl2, sono stati addizionati 0.016 g
(0.084 mmol) di CoCp2; la soluzione è stata quindi agitata per alcuni minuti. La
dissociazione della fosfina è stata osservata tramite analisi 31P{1H} NMR.
31P{1H} NMR (CH2Cl2 con capillare di C6D6, 293 K): δ (ppm) = 20.9 (s, P).

59
Alla soluzione contenente il cluster ridotto e la fosfina libera sono stati aggiunti 0.014
g (0.042 mmol) di [FeCp2][PF6]; la soluzione è stata quindi agitata per alcuni minuti.
Un prelievo è stato poi analizzato mediante 31P{1H} NMR.
31P{1H} NMR (CH2Cl2 con capillare di C6D6, 293 K): δ (ppm) = 20.9 (s, P), 76.0 (s,
P).
A questo punto, alla soluzione contenente sia il cluster ridotto che la fosfina libera
sono stati aggiunti altri 0.014 g (0.042 mmol) di [FeCp2][PF6]; la soluzione è stata
quindi agitata per alcuni minuti. La formazione del cluster iniziale 2 è stata osservata
tramite analisi 31P{1H} NMR.
31P{1H} NMR (CH2Cl2 con capillare di C6D6, 293 K): δ (ppm) = 76.0 (s, P).

60
APPENDICE I
Voltammetria ciclica La voltammetria ciclica (VC) è una tecnica sperimentale che può essere usata per una
valutazione preliminare delle proprietà redox di una specie nonché della
comunicazione elettronica tra centri remoti. Tale tecnica analitica, che fa parte delle
tecniche voltammetriche a scansione lineare, permette di misurare la corrente in
funzione del potenziale applicato ad un elettrodo stazionario, in condizioni di completa
polarizzazione. Il potenziale, sotto forma di onda triangolare, viene fatto variare
linearmente a velocità costante (in genere compresa tra 20 mV . s-1 e 100 V . s-1) da un
valore iniziale Ei ad uno finale Ef, dopo il quale, la direzione di scansione viene
invertita fino al raggiungimento del valore di potenziale iniziale Ei.
Un esperimento di voltammetria ciclica prevede l’uso di tre differenti elettrodi: un
elettrodo di lavoro, di materiale inerte (in genere Pt, Au o Hg), sul quale avvengono i
processi redox; un elettrodo di riferimento, dal potenziale noto, che funge da
riferimento interno; un contro-elettrodo (o elettrodo ausiliario), in genere costituito
da una griglia di Pt, il quale assicura che la corrente non passi all’elettrodo di
riferimento, che altrimenti potrebbe subire una variazione del suo potenziale. Il
voltammogramma (Fig. I.1) che viene registrato nel corso di un esperimento di
voltammetria ciclica, si presenta con una forma caratteristica a doppia onda: una per il
processo catodico ed una per quello anodico. I parametri più significativi da prendere
in considerazione sono (Fig. I.1):
- Epc: potenziale di picco catodico;
- Epa: potenziale di picco anodico;
- ipc: corrente al picco catodico;
- ipa: corrente al picco anodico.

61
L’analisi del profilo voltammetrico e della sua variazione in funzione della velocità di
scansione, consente la comprensione del tipo di meccanismo con cui avviene la
reazione nonché delle sue caratteristiche di reversibilità. Infatti, la reversibilità
elettrochimica e chimica sono strettamente correlate: un generico processo viene
definito elettrochimicamente reversibile quando la velocità del trasferimento
elettronico è superiore a quella del trasporto di massa; ciò consente il mantenimento
della coppia redox all’equilibrio nel corso della scansione di potenziale. Un processo
di questo tipo in genere comporta una riorganizzazione geometrica-strutturale modesta
in seguito al trasferimento elettronico: in tal caso si ha reversibilità chimica. Il profilo
voltammetrico per un processo di riduzione reversibile è riportato in Fig. I.2a. In
generale, in un processo reversibile le correnti anodica e catodica in corrispondenza
del picco sono circa uguali e la differenza tra i potenziali dei due picchi è uguale a
0.0592/n, dove n è il numero di elettroni coinvolti nella semireazione. Il potenziale di
semireazione (E0) è pari alla media aritmetica del potenziale al picco catodico e
anodico, ovvero (Epc + Epa)/2. Un processo elettrochimicamente irreversibile, invece,
ha luogo quando la velocità del trasferimento elettronico è inferiore a quella del
trasporto di massa; tale irreversibilità dipende dalla presenza di una barriera
d’attivazione elevata per il processo redox che può portare, in questi casi, alla
frammentazione della specie originaria ed alla formazione di nuovi composti. Il profilo
voltammetrico per un processo di riduzione irreversibile è riportato in Fig. I.2c.
Figura I.1 Voltammogramma.

62
Nei casi intermedi, nei quali le velocità dei due processi (trasferimento elettronico e
trasporto di massa) sono paragonabili, si parla di quasi-reversibilità: si tratta, infatti, di
processi caratterizzati da importanti riarrangiamenti molecolari che non comportano
necessariamente la decomposizione delle specie originarie (Fig. I.2b).
Relativamente ai parametri caratteristici di un voltammogramma, è possibile parlare
di processo reversibile quando:
la separazione tra Epc ed Epa (ΔEp), a 25 °C, è pari a 59/n mV (n = n° di e-
scambiati nel processo redox);
il potenziale del picco di andata (Ef) è indipendente dalla velocità di scansione;
il rapporto tra la corrente del picco di andata (ipf) e la radice della velocità di
scansione (v -1/2) rimane costante al variare della velocità di scansione;
il rapporto ipa / ipc rimane costantemente uguale ad 1.
Un processo irreversibile è, invece, caratterizzato dall’assenza del picco di ritorno e
dal fatto che il potenziale del picco di andata (Ef) varia in funzione della velocità di
scansione. In un processo quasi-reversibile all’aumentare della velocità di scansione si
ha una crescita del valore di ΔEp, mentre il rapporto ipa / ipc rimane solitamente pari ad
1.
La voltammetria consente la valutazione della comunicazione elettronica che si può
eventualmente instaurare fra centri metallici separati da leganti coniugati. Quando in
un complesso polinucleare i centri metallici interagiscono tra loro attraverso i leganti
Figura I.2 Profili voltammetrici relativi ad un processo di riduzione: a) reversibile,
b) quasi-reversibile, c) irreversibile.

63
o attraverso il legame diretto M-M, il potenziale di riduzione di ogni centro metallico
è influenzato dallo stato di riduzione del centro vicino. Il caso più semplice è quello di
un sistema dinucleare costituito da due atomi metallici identici, con gli stessi leganti e
lo stesso stato di ossidazione, connessi ad esempio da un legante coniugato a ponte. In
seguito ad un processo di riduzione monoelettronico nel quale viene ridotto uno dei
due centri metallici, il potenziale di riduzione del secondo centro metallico risulterà
spostato a valori più negativi a causa della presenza del primo centro metallico ridotto
(Fig. I.3).
Si hanno quindi due distinti processi di riduzione che in un esperimento di
voltammetria ciclica generano due onde di riduzione separate. In un sistema trinucleare
invece, saranno presenti tre distinti processi di riduzione poiché, dopo l’ingresso del
secondo elettrone, il terzo centro metallico risentirà dei due centri ridotti e di
conseguenza il suo potenziale di riduzione risulterà spostato a valori ancora più
negativi. Si riporta, a titolo di esempio, il profilo voltammetrico di un composto
dinucleare, contenente due centri metallici (M) connessi da uno spaziatore organico,
in assenza ed in presenza di comunicazione elettronica (Fig. I.4).
Figura I.4 Profilo voltammetrico relativo ad un composto dinucleare in assenza (a) ed in
presenza (b) di comunicazione elettronica.
Figura I.3 Sistema dinucleare connesso mediante uno spaziatore coniugato: i due atomi metallici
risentono uno dello stato di ossidazione dell’altro.

64
L’entità dell’interazione fra i centri metallici viene in genere valutata attraverso la
misura della costante di equilibrio Kc (equilibrio di comproporzione). In Fig. I.5 si
riporta l’esempio del complesso dinucleare I, dal quale viene generato il complesso a
valenza mista VM per rimozione di un elettrone da uno dei due centri metallici; dal
complesso VM si genera poi il complesso II a seguito di una successiva ossidazione
monoelettronica.
Le specie I, VM e II sono correlate mediante quello che si definisce equilibrio di
comproporzione mostrato nello schema di Fig. I.6, la cui costante Kc è funzione della
differenza tra i potenziali dei due processi redox riportati in Fig. I.5 (∆𝐸° = 𝐸2 ° − 𝐸1
°).
La concentrazione della specie a valenza mista VM tende a zero al diminuire del ΔE°
e la comunicazione elettronica è efficiente quando si osservano valori elevati di ΔE°
e, quindi, della Kc. In questi casi è in genere possibile isolare la specie VM anche per
via chimica [83]. La Kc può essere definita in accordo con la seguente espressione:
𝐾𝑐 = 𝑒(∆𝐸°𝐹
𝑅𝑇) (I.1).
Ad ogni modo, un’efficiente comunicazione elettronica lungo una struttura è il
risultato di molti fattori e dipende non solo dalle caratteristiche elettroniche del
frammento organico e del frammento metallico, ma anche dal modo con cui questi
interagiscono fra di loro.
Figura I.5 Equilibri redox di una generica specie Mn-spaziatore-Mn.
Figura I.6 Equilibrio di comproporzione per una generica specie Mn-spaziatore-Mn.

65
APPENDICE II
Spettroelettrochimica Nel corso degli ultimi decenni la spettroelettrochimica (SEC) si è affermata come una
tecnica efficace nel monitoraggio di processi elettrochimici ed utile nella raccolta di
dati identificativi dei prodotti elettrogenerati. D’altronde, sebbene le tradizionali
tecniche analitiche elettrochimiche siano metodi eccellenti per la determinazione della
concentrazione di una specie, per la raccolta di dati energetici sotto forma di potenziali
redox o per la comprensione di meccanismi di reazione mediante analisi cinetiche, tali
tecniche, da sole, non sono sufficienti per l’identificazione di specie ignote formatesi
come intermedi o prodotti nel corso di una reazione redox. Pertanto, la possibilità di
poter utilizzare ulteriori e più specifiche caratteristiche fisiche delle molecole al fine
di monitorare i processi elettrodici, sia in condizioni dinamiche che di equilibrio, ha
generato enormi sforzi nello sviluppo delle tecniche spettroelettrochimiche [84].
L’impulso decisivo nell’affermazione della SEC è stato fornito dalla disponibilità, a
partire dalla fine degli anni ’50, di elettrodi otticamente trasparenti (OTE) mediante i
quali è stato possibile effettuare misure spettroscopiche direttamente attraverso
l’elettrodo. Tra i materiali più usati per avere trasparenza ottica nelle regioni IR ed
UV-Vis vi è l’ossido di stagno drogato con indio; più recentemente sono stati messi a
punto elettrodi costituiti da film di diamante drogati con boro, i quali consentono la
scansione di ampi intervalli di potenziale garantendo inerzia in ambienti chimicamente
aggressivi e biocompatibilità. Una strategia alternativa all’uso di elettrodi trasparenti
è data da conduttori nobili in forma di griglie o garze sottili [85].
Le celle impiegate comunemente sino alla fine degli anni ’60 avevano tempi di risposta
variabili da circa 10 s fino anche ad alcuni minuti: ciò comportava che processi
chimicamente reversibili sulla scala dei tempi della voltammetria ciclica potevano
risultare irreversibili nel lungo intervallo necessario all’acquisizione della risposta in
un esperimento spettroelettrochimico. Il tempo di risposta è stato tuttavia ridotto grazie
all’introduzione di celle, come quella schematizzata in Fig. II.1, in cui lo spessore dello
strato di elettrolita attorno all’elettrodo di lavoro è più piccolo.

66
Ad ogni modo, nel corso degli anni, sono state messe a punto numerose configurazioni
per le celle, in modo che queste possano risultare adeguate ed in grado di rispondere a
diversi requisiti sperimentali, come ad esempio l’assenza di aria ed umidità, la capacità
di condurre misure a differenti temperature, la possibilità di un controllo molto fine
del potenziale applicato, la geometria variabile che ne permette l’uso con diversi tipi
di spettrometro [86]. Infatti, i metodi spettroscopici che sono applicati congiuntamente
all’elettrochimica sono numerosi e includono le comuni spettroscopie di assorbimento
nelle regioni UV-Vis ed IR, la spettroscopia Raman, le risonanze magnetiche NMR ed
ESR, la spettroscopia di assorbimento dei raggi X, la luminescenza nell’UV-Vis.
La spettroelettrochimica viene utilizzata per motivi che vanno oltre la semplice
raccolta di dati spettroscopici delle specie elettrogenerate; tra questi, ad esempio, si
possono annoverare: l’identificazione del sito realmente coinvolto nel processo di
trasferimento elettronico all’interno di una molecola con due o più siti redox attivi con
potenziali d’ossidazione o riduzione simili, ma proprietà differenti delle rispettive
forme ossidate o ridotte; l’ottenimento di informazioni relativamente all’entità
dell’accoppiamento elettronico sussistente tra unità redox legate attraverso spaziatori.
Tra le problematiche, invece, associate all’analisi spettroelettrochimica vanno
ricordate la difficoltà nel rilasciare o mantenere un’uniforme distribuzione corrente-
potenziale all’interno di una cella a strato sottile nella quale si hanno, spesso,
separazioni notevoli tra gli elettrodi, la richiesta in alcuni casi di un tempo di elettrolisi
Figura II.1 Schema di una cella OTTLE (Optically Trasparent Thin-Layer Electrode)
per misure nelle regioni UV-Vis e IR.

67
lungo per convertire tutto il reagente di partenza e la possibilità che l’elettrolita di
supporto od il solvente interferiscano nell’identificazione degli intermedi generati
[87]. Nonostante ciò la spettroelettrochimica rispetto, ad esempio, all’ossidazione
chimica elimina il problema del trasferimento del campione nella cella spettroscopica,
consente di individuare ed acquisire informazioni strutturali anche su intermedi con
tempo di vita breve e soprattutto consente la diretta correlazione di dati spettroscopici
ed elettrochimici in funzione del tempo e del potenziale [88].

68
BIBLIOGRAFIA
[1] F. A. Cotton, Inorg. Chem., 1964, 3, 1217.
[2] G. González-Moraga, Cluster Chemistry, Springer-Verlag Berlin Heidelberg GmbH, 1993.
[3] R. H. Crabtree, The Organometallic Chemistry of Transition Metals, Wiley-Interscience, 4° ed.,
2005.
[4] a) J. A. Bertrand, F. A. Cotton, W. A. Dollase, J. Am. Chem. Soc., 1963, 85, 1349; b) J. A. Bertrand,
F. A. Cotton, W. A. Dollase, Inorg. Chem., 1963, 2, 1166; c) F. A. Cotton, N. F. Curtis, C. B. Harris et
al., 1964, 145, 1305; d) F. A. Cotton, Inorg. Chem., 1965, 4, 334; e) F. A. Cotton, C. B. Harris, Inorg.
Chem., 1965, 4, 330.
[5] a) D. M. P. Mingos, D. J. Wales, Introduction to Cluster Chemistry, Prentice Hall Advanced
Reference Series, inorganic and organometallic chemistry series, 1990; b) P. Braunstein, L. A. Oro, P.
R. Raithby, Metal Clusters in Chemistry, Wiley-VCH, 1999.
[6] D. M. P. Mingos, D. J. Wales, J. Organomet. Chem., 1990, 349, 679.
[7] I. Langmuir, Types of Valence, Science, 1921, 54, 59.
[8] a) K. Wade, J. Chem. Soc. Chem. Commun., 1971, 15, 792; b) R. Manson, K. M. Thomas, D. M. P.
Mingos, J. Am. Chem. Soc., 1973, 95, 3802; c) D. M. P. Mingos, Polyhedron, 1984, 3, 1289; d) K. P.
Hall, D. M. P. Mingos, Prog. Inorg. Chem., 1984, 32, 237; e) D. M. P. Mingos, M. J. Watson, Adv.
Inorg. Chem., 1992, 39, 327.
[9] J. Farges, M. F. de Feraudy, B. Raoult, G. Torchet, J. Chem. Phys., 1986, 84, 3491.
[10] a) F. Demartin, C. Femoni, M. C. Iapalucci, G. Longoni, P. Macchi, Angew. Chem. Int. Edit., 1999,
38, 531; b) C. Femoni, M. C. Iapalucci, G. Longoni, P. H. Svensson, J. Wolowska, Angew. Chem. Int.
Edit., 2000, 39, 1635; c) C. Femoni, M. C. Iapalucci, G. Longoni, P. H. Svensson, P. Zanello, F. de
Biani, Chemistry – A European Journal, 2004, 10, 2318; d) F. Demartin, F. de Biani, C. Femoni, M. C.
Iapalucci, G. Longoni, P. Macchi, P. Zanello, Journal Cluster of Science, 2001, 12, 61; e) C. Femoni,
M. C. Iapalucci, G. Longoni, P. H. Svensson, Chem. Commun., 2004, 0, 2274.
[11] F. de Biani, C. Femoni, M. C. Iapalucci, G. Longoni, P. Zanello, A. Ceriotti, Inorg. Chem., 1999,
38, 3721.
[12] a) T. H. Lee, J. I. González, J. Zheng, M. R. Dickson, Accounts of Chemical Research, 2005, 38,
534; b) D. Collini, C. Femoni, M. C. Iapalucci, G. Longoni, P. H. Svensson, P. Zanello, Angew. Chem.
Int. Edit., 2002, 41, 3685; c) C. Femoni, M. C. Iapalucci, F. Kaswalder, G. Longoni, S. Zacchini,
Coordination Chemistry Reviews, 2006, 250, 1580.
[13] a) N. T. Tran, D. R. Powell, L. F. Dahl, Angew. Chem. Int. Edit., 2000, 39, 4121; b) N. T. Tran, M.
Kawano, L. F. Dahl, J. Chem. Soc. Dalton Trans., 2001, 0, 2731; c) M. Kawano, J. W. Bacon, C. F.
Campana, B. E. Winger, J. D. Dudek, S. A. Sirchio, S. L. Scruggs, U. Geiser, L. F. Dahl, Inorg. Chem.,
2001, 40, 2554; d) G. J. Lewis, J. D. Roth, R. A. Montag, L. K. Safford, X. Gao, S. C. Chang, L. F.
Dahl, M. J. Weaver, J. Am. Chem. Soc., 1990, 112, 2831; e) E. G. Mednikov, M. C. Jewell, L. F. Dahl,
J. Am. Chem. Soc., 2007, 129, 11619.

69
[14] a) A. Schnepf, H. Schnöckel, Angew. Chem. Int. Edit, 2002, 41, 3532; b) A. Ecker, E. Weckert, H.
Schnöckel, Nature, 1997, 387, 379; c) A. Schnepf, R. Köppe, E. Weckert, H. Schnöckel, Chemistry –
A European Journal, 2004, 10, 1977; d) A. Schnepf, H. Schnöckel, Angew. Chem. Int. Edit., 2001, 40,
711; e) H. Schnöckel, Dalton Trans., 2005, 0, 3131.
[15] a) F. de Biani, C. Femoni, M. C. Iapalucci, G. Longoni, P. Zanello, A. Cerotti, Inorg. Chem., 1999,
28, 179; b) G. Schmid, Chem. Rev., 1992, 92, 1709.
[16] E. L. Muetterties, Chem. Soc. Rev., 1982, 11, 283.
[17] W. H. Brown, C. S. Foote, B. L. Iverson, Chimica organica, EdiSES, 3° ed., 2006.
[18] F. S. Arimoto, A. C. Haven, Jr., J. Am. Chem. Soc., 1955, 77, 6295.
[19] a) T. Yamamoto, K.-I. Sanechika, A. Yamamoto, M. Katada, I. Motoyama, H. Sano, Inorg. Chim.
Acta, 1983, 73, 75; b) T. Morikita, T. Yamamoto, J. Organomet. Chem., 2001, 637-639, 809-812.
[20] a) R. Yoshida, T. Takahashi, T. Yamaguchi, H. Ichijo, Adv. Mater., 1997, 9, 175; b) A. C.
Arsenault, H. Mìguez, V. Kitaev, G. A. Ozin, I. Manners, Adv. Mater., 2003, 15, 503.
[21] a) D. J. Caruana, A. Heller, J. Am. Chem. Soc., 1999, 121, 769; b) M. Albrecht, G. Van Koten, Adv.
Mater., 1999, 11, 171; c) B. Wang, M. R. Wasielewski, J. Am. Chem. Soc., 1997, 19, 12.
[22] a) A. N. Ajjou, H. Alper, J. Am. Chem. Soc., 1998, 120, 1466; b) T. J. Peckham, P. Nguyen, S. C.
Bourke, Q. Wang, D. G. Harrison, P. Zoricak, C. Russell, L. M. Liable-Sands, A. L. Rheingold, A. J.
Lough, I. Manners, Organometallics, 2001, 20, 3035; c) C. Kollner, B. Pugin, A. Togni, J. Am. Chem.
Soc., 1998, 120, 10274.
[23] a) K. D. Ley, C. Ed Whittle, M. D. Bartberger, K. S. Schanze, J. Am Chem. Soc., 1997, 119, 3423;
b) C. T. Wong, W. K. Chan, Adv. Mater., 1999, 11, 455.
[24] B. J. Holliday and T. M. Swager, Chem. Commun., 2005, 23.
[25] A. M. Bradford, E. Kristof, M. Rashidi, D.-S. Yang, N. C. Payne, R. J. Puddephatt, Inorg. Chem.,
1994, 33, 2355.
[26] B. F. G. Johnson, K. M. Sanderson, D. S. Shepard, D. Ozkaya, W. Zhou, H. Ahmed, M. D. R.
Thomas, L. Gladden, M. Mantle, Chem. Comm., 2000, 8, 1317.
[27] N. T. Lucas, M. G. Humprey, A. D. Rae, Macromolecules, 2001, 34, 6188.
[28] F. Wang, Y.-H. Lay, M. Y. Han, Org. Lett., 2003, 5, 4791.
[29] a) H. Kim, D.-S. Park, Y.-B. Shim, S. C. Shin, J. Organomet. Chem., 2000, 608, 133; b) C. W.
Allen, M. J. Bahadur, Inorg. Organomet. Polym., 1998, 8, 23; c) B. S. Kang, D. H. Kim, T. S. Jung, E.
K. Jang, Y. Pak, S. C. Shin, D.-S. Park, Y.-B. Shim, Synth. Metals, 1999, 105, 9; d) H. D. Selby, B. K.
Roland, Z. Zheng, Acc. Chem. Res., 2003, 36, 933.
[30] B. K. Roland, W. H. Flora, M. D. Carducci, N. R. Armstrong, Z. Zheng, J. Clust. Sci., 2003, 14,
449.
[31] W. Y. Chan, S. B. Clendenning, A. Berenbaum, A. J. Lough, S. Aouba, H. E. Ruda, I. Manners, J.
Am. Chem. Soc., 2005, 127, 1765.
[32] H. Krogman, Angew. Chem. Int. Ed., 1969, 8, 35.
[33] a) D. Vrbani´c, M. Remˇskar, A. Jesih, A. Mrzel, P. Umek, M. Ponikvar, B. Janˇcar, A. Meden, B.
Novosel, S. Pejovnik, P. Venturini, J. C. Coleman, D. Mihailovic, Nanotechnology, 2004, 15, 635; b)

70
D. Mihailovic, Progress in Materials Science, 2009, 54, 309; c) J. M. Tarascon, G. W. Hull, F. J. Di
Salvo, Mater. Res. Bull., 1984, 19, 915.
[34] J. F. Berry, F. A. Cotton, C. A. Murillo, Dalton Trans., 2003, 3015.
[35] a) T. Murahashi, T. Nagai, T. Okuno, T. Matsutani, H. Kurosawa, Chem. Comm., 2000, 1689; b)
T. Murahashi, E. mochizuki, Y. Kai, H. Kurosawa, J. Am. Chem. Soc., 1999, 121, 10660.
[36] a) M. Benito, O. Rossell, M. Seco, G. Segalés, Organometallics, 1999, 18, 5191; b) J. R. Aranzaes,
C. Berlin, D. Astruc, Angew. Chem. Int. Ed., 2006, 45, 132; c) N. Feeder, J. Geng, P. G. Goh, B. F. G.
Johnson, C. M. Martin, D. S. Shepard, W. Zhou, Angew. Chem. Int. Ed., 2000, 39, 1661.
[37] D. Méry, L. Plault, C. Ornelas, J. Ruiz, S. Nlate, D. Astruc, J. C. Blaise, J. Rodrigues, S. Cordier,
K. Kirakci, C. Perrin, Inorg. Chem., 2006, 45, 1156.
[38] a) R. Mason, A. I. M. Rae, J. Chem. Soc. A, 1968, 778; b) M. R. Churchill, B. G. DeBoer,
Inorg.Chem. 1977, 16, 878; c) J. A. Connor, Top. Curr. Chem. 1977, 71, 71.
[39] a) P. Chini, J. Organomet. Chem. 1980, 200, 37; b) J. C. Calabrese, L. F. Dahl, A. Cavalieri, P.
Chini, G. Longoni, S. Martinengo, J. Am. Chem. Soc., 1974, 96, 2614.
[40] a) P. Leoni, S. Manetti, M. Pasquali, A. Albinati, Inorg. Chem., 1996, 35, 6045; b) P. Leoni, F.
Marchetti, L. Marchetti, M. Pasquali, S. Quaglierini, Angew. Chem. Int. Ed., 2001, 40, 3617; c) S. K.
Mohapatra, Tesi di Dottorato, Università di Pisa, 2008.
[41] a) P. Leoni, F. Marchetti, L. Marchetti, M. Pasquali, Chem. Commun., 2003, 2372; b) A. Albinati,
P. Leoni, L. Marchetti, S. Rizzato, Angew. Chem., Int. Ed., 2003, 42, 5990.
[42] P. Leoni, L. Marchetti, S. K. Mohapatra, G. Ruggeri, L. Ricci, Organometallics, 2006, 25, 4226.
[43] F. Fabrizi de Biani, A. Ienco, F. Laschi, P. Leoni, F. Marchetti, L. Marchetti, C. Mealli, P. Zanello,
J. Am. Chem. Soc., 2005, 127, 3076.
[44] A. Albinati, F. Fabrizi de Biani, P. Leoni, L. Marchetti, M. Pasquali, S. Rizzato, P. Zanello, Angew.
Chem. Int. Ed., 2005, 44, 5701.
[45] a) A. M. Shaw, Astrochemistry: from astronomy to astrobiology, Ed. Wiley, 1° ed., 2006; b) G. L.
Miessler, D. A. Tarr, Inorganic chemistry, Ed. Pearson Prentice Hall, 2010.
[46] A. L. Barra, A. Caneschi, A. Cornia, F. Fabrizi de Biani, D. Gatteschi, C. Sangregorio, R. Sessoli,
L. Sorace, J. Am. Chem. Soc., 1999, 121, 5302.
[47] a) H.-C. Boettcher, A. Krug, H. Hartung, Polyhedron, 1995, 14, 901; b) B. Walther, H. Hartung,
S. Bambirra, A. Krug, H.-C. Boettcher, Organometallics, 1994, 13, 172; c) C. P. Horwitz, D. F. Shriver,
Organometallics, 1984, 3, 756; d) C. Goh, B. M. Segal, J. Huang, J. R. Long, R. H. Holm, J. Am. Chem.
Soc., 1996, 118, 11844; e) C. A. Goddard, J. R. Long, R. H. Holm, Inorg. Chem., 1996, 35, 4347.
[48] E. H. Braye, L. F. Dahl, W. Hübel, D. L. J. Wampler, J. Am. Chem. Soc., 1962, 84, 4633.
[49] a) M. Tachikawa, R. L. Geerts, E. L. Muetterties, J. Organomet. Chem., 1981, 213, 11; b) J. S.
Bradley, E. W. Hill, G. B. Ansell, M. A. Modrick, Organometallics, 1982, 1, 1634.
[50] B. F. G. Johnson, J. Lewis, J. N. Nicholls, J. Puga, P. R. Raithby, M. J. Rosales, M. McPartlin, W.
Clegg, J. Chem. Soc. Dalton Trans., 1983, 277.
[51] a) A. J. Poë, Y. Zheng, Inorg. Chim. Acta, 1996, 252, 311; b) A. J. Poë, D. H. Farrar, Y. Zheng, J.
Am. Chem. Soc., 1992, 114, 5146; c) A. Gourdon, Y. Jeannin, J. Organomet. Chem., 1990, 388, 195; d)
C. G. Cooke, M. Mays, J. Organomet. Chem., 1975, 88, 231.

71
[52] M. Ferrer, R. Reina, O. Rossell, M. Seco, Coord. Chem. Rev., 1999, 193-195, 619.
[53] R. Della Pergola, A. Sironi, L. Garlaschelli, D. Strumolo, C. Manassero, M. Manassero, S. Fedi, P.
Zanello, F. Kaswalder, S. Zacchini, Inorg. Chim. Acta, 2010, 363, 586.
[54] R. D. Adams, B. Captain, W. Fu, J. Cluster Sci., 2001, 12, 303.
[55] a) O. Rossel, M. Seco, G. Segalés, S. Alvarez, M. A. Pellinghelli, A. Tiripicchio, D. de Montauzon,
Organometallics, 1997, 16, 236; b) R. Reina, O. Riba, O. Rossell, M. Seco, P. Gómez-Sal, A. Martín,
D. de Montauzon, A. Mari, Organometallics, 1998, 17, 4127; c) J. Camats, R. Reina, O. Riba, O.
Rossell, M. Seco, P. Gómez-Sal, A. Martín, D. de Montauzon, Organometallics, 2000, 19, 3316.
[56] a) C. E. Sumner Jr., J. A. Collier, R. Pettit, Organometallics, 1982, 1, 1350; b) E. H. Braye, W.
Hübel, M. D. Rausch, T. M. Wallace, Inorg. Synth., 1966, 8, 178; c) W. Mcfarlane, G. Wilkinson, W.
Hübel, Inorg. Synth., 1966, 8, 181; d) R. L. Holliday, L. C. Roof, B. Hargus, D. M. Smith, P. T. Wood,
W. T. Pennington, J. W. Kolis, Inorg. Chem., 1995, 34, 4392.
[57] a) H.-C. Boettcher, H. Hartung, A. Krug, Zeitschrift fuer Naturforschung B, 1995, 50, 1175; b) R.
D. Moulton, D. J. Chandler, A. M. Arif, R. A. Jones, A. J. Bard, J. Am. Chem. Soc., 1988, 110, 5714;
c) A. M. Arif, D. J. Chandler, R. A. Jones, J. Coord. Chem., 1988, 17, 45; d) A. M. Arif, D. J. Chandler,
R. A. Jones, Organometallics, 1987, 6, 506; e) M. R. Adams, J. Gallucci, A. Wojcicki, G. J. Long,
Inorg. Chem., 1992, 31, 2; f) H.-C. Boettcher, M. Graf, K. Merzweiler, J. Organomet. Chem., 1997,
534, 43; g) H.-C. Boettcher, M. Graf, K. Merzweiler, J. Organomet. Chem., 1997, 531, 107; h) V. D.
Patel, A. A. Cherkas, D. Nucciarone, N. J. Taylor, A. J. Carty, Organometallics, 1985, 4, 1792.
[58] a) E. W. Hill, J. S. Bradley, J. Cassidy, K. H. Whitmire, Inorg. Synth., 1990, 27, 182; b) M. R.
Churchill, J. Wormald, J. Chem. Soc. Dalton Trans., 1974, 2410.
[59] O. Rossel, M. Seco, G. Segalés, M. A. Pellinghelli, A. Tiripicchio, J. Organomet. Chem., 1998,
571, 123.
[60] a) I. O. Koshevoy, M. Haukka, T. A. Pakkanen, S. P. Tunik, Dalton Trans., 2006, 5641; b) W.
Wang, P. J. Low, A. J. Carty, E. Sappa, G. Gervasio, C. Mealli, A. Ienco, E. Perez-Carreño, Inorg.
Chem., 2000, 39, 998; c) M. P. Cifuentes, M. G. Humphrey, B. W. Skelton, A. H. White,
Organometallics, 1993, 12, 4272; d) S. Chan, S.-M. Lee, Z. Lin, W.-T. Wong, J. Organomet Chem.,
1996, 510, 219; e) M. I. Bruce, J. R. Rodgers, M. R. Snow, F. S. Wong, J. Chem. Soc., Chem. Commun.,
1980, 1285.
[61] M. L. H. Green, A. Hamnet, J. Qin, P. Baird, J. A. Bandy, K. Prout, E. Marseglia, S. D. Obertelli,
J. Chem. Soc. Chem., Commun., 1987, 1811.
[62] J. Podlahová, J. Podlaha, A. Jegorov, J. Hašek, J. Organomet. Chem., 1989, 359, 401.
[63] M. I. Bruce, J. R. Rodgers, M. R. Snow, F. S. Wong, J. Organomet Chem., 1982, 240, 299.
[64] F. Fabrizi de Biani, A. Ienco, F. Laschi, P. Leoni, F. Marchetti, L. Marchetti, C. Mealli, P. Zanello,
J. Am. Chem. Soc., 2005, 127, 3076.
[65] a) D.A. Roberts, G.L. Geoffrey, Comprehensive Organometallic Chemistry, Pergamon Press,
Oxford, 6, 763, Ed. G. Wilkinson, G. Stone, E.W. Abel, 1982; b) G. Wilkinson, G. Stone, E.W. Abel,
Comprehensive Organometallic Chemistry II, Pergamon Press, Oxford, 10, 1995.
[66] R. Della Pergola, M. Bruschi, A. Sironi, M. Manassero, C. Manassero, D. Strumolo, S. Fedi, P.
Zanello, Dalton Trans., 2011, 5464.
[67] R. Della Pergola, A. Sironi, C. Manassero, M. Manassero, Eur. J. Inorg. Chem., 2009, 4618.

72
[68] a) T. Nakajima, H. Konomoto, H. Ogawa, Y. Wakatsuki, J. Organomet. Chem., 2007, 692, 5071;
b) U. Brand, J. L. Coffer, T. J. Henly, S. R. Wilson, J. R. Shapley, Inorg. Chem., 1997, 36, 3386; c) R.
Della Pergola, A. Sironi, C. Manassero, M. Manassero, Organometallics, 2010, 29, 5885; d) M. Shieh,
M.-H. Hsu, W.-S. Sheu, L.-F. Jang, S.-F. Lin, Y.-Y. Chu, C.-Y. Miu, Y.-W. Lai, H.-L. Liu, J. L. Her,
Chem. Eur. J., 2007, 13, 6605; e) M. Shieh, C.-Y. Huang, C.-J. Lee, K.-J. Hsing, Y.-W. Li, Y.-Y. Chu,
W.-T. Jhu, Polyhedron, 2013, 52, 879.
[69] E. E. Chufán, S. C. Puiu, K. D. Karlin, Acc. Chem. Res., 2007, 40, 563.
[70] a) B. Wu, L. Tian, H. Xiang, Z. Zhang, Y.-W. Li, Catal. Lett., 2005, 102, 211; b) P. Braunstein, G.
Clerc, X. Morise, New J. Chem., 2003, 27, 68.
[71] K. Albert, K. M. Neyman, G. Pacchioni, N. Rösch, Inorg. Chem., 1996, 35, 7370.
[72] T. J. Henly, J. R. Shapley, Organometallics, 1989, 8, 2729.
[73] a) R. Della Pergola, F. Demartin, L. Garlaschelli, M. Manassero, S. Martinengo, N. Masciocchi,
M. Sansoni, Organometallics, 1991, 10, 2239; b) A. Ceriotti, R. Della Pergola, L. Garlaschelli, M.
Manassero, N. Masciocchi, Organometallics, 1995, 14, 186.
[74] C. J. Jones, Chem. Soc. Rev., 1998, 27, 289.
[75] D. A. Skoog, F. J. Holler, S. R. Crouch, Chimica analitica strumentale, EdiSES, II ed., 2009.
[76] a) P. Zanello, Coord. Chem. Rev., 1988, 83, 199; b) F. Fabrizi de Biani, C. Femoni, M. C. Iapalucci,
G. Longoni, P. Zanello, A. Cerotti, Inorg. Chem., 1999, 38, 3721.
[77] a) M. K. Johnson, R. E. Duderstadt, E. C. Duin, Adv. Inorg. Chem., 1999, 47, 1; b) R. H. Holm,
Adv. Inorg. Chem., 1992, 38, 1.
[78] A. Gourdon, Y. Jeannin, J. Organomet. Chem., 1985, 290, 199.
[79] J. Rimmelin, P. Lemoine, M. Gross, R. Mathieu, D. De Montauzon, J. Organomet. Chem., 1986,
309, 355.
[80] C. Cavazza, F. Fabrizi de Biani, T. Funaioli, P. Leoni, F. Marchetti, L. Marchetti, P. Zanello, Inorg.
Chem., 2009, 48, 1385.
[81] W. Kaim, A. Klein, Spectroelectrochemistry, Royal Society of Chemistry, 2008.
[82] a) H. Hoffmann, P. Schellenbeck, Chem. Ber., 1966, 99, 1134; b) P. Leoni, M. Sommovigo, M.
Pasquali, P. Sabatino, D. Braga, J. Organomet. Chem., 1992, 423, 263.
[83] a) D. Astruc, Electron Transfer and Radical Processes in Transition-Metal Chemistry, Wiley-VCH
Edition, 1995; b) P. Zanello, Inorganic Electrochemistry: Theory, Practice and Application, The Royal
Society of Chemistry, 2003.
[84] R. J. Gale, Spectroelectrochemistry: Theory and Practice, Springer Edition, 1988.
[85] J. Stotter, J. Zak, Z. Behler, Y. Show, G.-M. Swain, Anal. Chem., 2002, 74, 5924.
[86] W. Kaim, J. Fiedler, Chem. Soc. Rev., 2009, 38, 3373.
[87] P. B. Graham, D. J. Curran, Anal. Chem., 1992, 64, 2688.
[88] N. G. Connelly, W. E. Geiger, Chem. Rev., 1996, 96, 877.

73
“…vincere la materia è comprenderla, e comprendere la materia è necessario per comprendere l’universo e noi
stessi…” Da ‘Ferro’ di Primo Levi
Ai miei genitori, a Valeria.


















