
Capitolo 1 IL CARBONE FOSSILE Il carbone fossile · La composizione dei diversi tipi di carbone è...
39
1 Capitolo 1 IL CARBONE FOSSILE Il carbone fossile Il carbone fossile è il risultato della degradazione indotta da agenti biologici, chimici e fisici di depositi organici, nel corso delle ere geologiche. Il grado di conversione (fossilizzazione) subito dalle sostanze originarie è in relazione col contenuto di carbonio e serve per una classificazione dei carboni fossili per tipologia. La composizione dei diversi tipi di carbone è riportata nella Tabella 1. Tabella 1 Composizione dei carboni fossili Tipo Composizione (% in peso)* Potere cal. C H O N Volatili Umidità Kcal/g Torba 45-60 3,5-6,8 20-45 0,8-3,0 45-75 70-90 4,1-5,3 Lignite 60-75 4,5-5,5 17-35 0,8-2,1 45-60 30-50 6,7-7,2 Litantrace 75-92 4,0-5,5 3,0-20 0,7-2,0 11-50 1,0-20 6,9-8,8 Antracite 92-95 2,9-4,0 2,0-3,0 0,5-2,0 3,5-10 1,5-3,5 8,6-8,9 * Calcolata sul carbone secco (con l’eccezione del valore di umidità). Le denominazioni usate in Italia per i carboni fossili (antraciti e litantraci), classificati secondo la normativa stabilita nel 1956 dall’apposito Comitato della Comunità Economica Europea come mostrato nelle prime tre colonne, sono riportate nella Tabella 2. Nella stessa Tabella sono anche riportate le denominazioni corrispondenti usate negli Stati Uniti. Tabella 2 Classificazione dei carboni fossili Classe n° Sost. volatili (%) Potere calor. (Kcal/g) ITALIA U.S.A. 0 0-3 antraciti speciali meta-anthracite 1A 3-6,5 antraciti comuni anthracite 1B 6,5-10 2 10-14 carboni magri semianthracite 3 14-20 carb. semigrassi low volatile bituminous 4 20-28 carboni grassi corta fiamma medium volatile bituminous 5 28-33 carboni grassi media fiamma high volatile bituminous A 6 33-40 7,75-8,47 carboni da gas 7 32-44 7,20-7,75 carboni grassi da vapore high volatile bituminous B 8 34-46 6,12-7,20 carboni secchi high volatile bituminous C 9 36-48 <6,12 subbituminous I più comuni gruppi funzionali ossigenati contenuti nel carbone fossile (si noti, in Tabella 1, che il tenore di ossigeno varia in modo molto forte nell’intervallo dei litantraci ed è molto basso per le antraciti) sono quelli carbossilici, carbonilici, ossidrilici e metossilici. Nei carboni più vecchi, l’ossigeno è presente come gruppi fenolici e carbonilici coniugati (chinonici).I gruppi contenenti azoto sono nitrili aromatici, piridine, carbazoli, chinoline e pirroli. Lo zolfo è principalmente presente come tioli, tioeteri dialchilici o aril-alchilici, gruppi tiofenici e disolfuri. Talvolta può essere presente anche zolfo elementare. Lo schema della struttura di un litantrace è dato in Figura 1.
Transcript of Capitolo 1 IL CARBONE FOSSILE Il carbone fossile · La composizione dei diversi tipi di carbone è...
1
Capitolo 1
IL CARBONE FOSSILE Il carbone fossile
Il carbone fossile è il risultato della degradazione indotta da agenti biologici, chimici e fisici di depositi organici, nel corso delle ere geologiche. Il grado di conversione (fossilizzazione) subito dalle sostanze originarie è in relazione col contenuto di carbonio e serve per una classificazione dei carboni fossili per tipologia. La composizione dei diversi tipi di carbone è riportata nella Tabella 1.
Tabella 1
Composizione dei carboni fossili
Tipo Composizione (% in peso)* Potere cal.
C H O N Volatili Umidità Kcal/g
Torba 45-60 3,5-6,8 20-45 0,8-3,0 45-75 70-90 4,1-5,3 Lignite 60-75 4,5-5,5 17-35 0,8-2,1 45-60 30-50 6,7-7,2 Litantrace 75-92 4,0-5,5 3,0-20 0,7-2,0 11-50 1,0-20 6,9-8,8 Antracite 92-95 2,9-4,0 2,0-3,0 0,5-2,0 3,5-10 1,5-3,5 8,6-8,9 * Calcolata sul carbone secco (con l’eccezione del valore di umidità).
Le denominazioni usate in Italia per i carboni fossili (antraciti e litantraci), classificati secondo la normativa stabilita nel 1956 dall’apposito Comitato della Comunità Economica Europea come mostrato nelle prime tre colonne, sono riportate nella Tabella 2. Nella stessa Tabella sono anche riportate le denominazioni corrispondenti usate negli Stati Uniti.
Tabella 2
Classificazione dei carboni fossili
Classe
n°
Sost. volatili
(%)
Potere calor.
(Kcal/g) ITALIA U.S.A.
0 0-3 antraciti speciali meta-anthracite 1A 3-6,5 antraciti comuni anthracite 1B 6,5-10 2 10-14 carboni magri semianthracite
3 14-20 carb. semigrassi low volatile bituminous
4 20-28 carboni grassi corta fiamma
medium volatile bituminous
5 28-33 carboni grassi media fiamma
high volatile bituminous A
6 33-40 7,75-8,47 carboni da gas
7 32-44 7,20-7,75 carboni grassi
da vapore high volatile bituminous B
8 34-46 6,12-7,20 carboni secchi high volatile bituminous C
9 36-48 <6,12 subbituminous
I più comuni gruppi funzionali ossigenati contenuti nel carbone fossile (si noti, in Tabella 1, che il tenore di ossigeno varia in modo molto forte nell’intervallo dei litantraci ed è molto basso per le antraciti) sono quelli carbossilici, carbonilici, ossidrilici e metossilici. Nei carboni più vecchi, l’ossigeno è presente come gruppi fenolici e carbonilici coniugati (chinonici).I gruppi contenenti azoto sono nitrili aromatici, piridine, carbazoli, chinoline e pirroli. Lo zolfo è principalmente presente come tioli, tioeteri dialchilici o aril-alchilici, gruppi tiofenici e disolfuri. Talvolta può essere presente anche zolfo elementare. Lo schema della struttura di un litantrace è dato in Figura 1.

2
Con riferimento alle utilizzazioni dei carboni fossili, le proprietà che assumono maggiore importanza sono il tipo di carbone, secondo la classificazione data in Tabella 2, generalmente legato all’età e coinvolgente molte proprietà tra le quali il tenore di sostanze volatili, il potere calorifico e la tendenza al rammollimento ed alla cokificazione. Altre proprietà importanti sono la facilità di macinazione, il contenuto di ceneri e quello di zolfo. La maggior parte del carbone fossile prodotto è impiegato come combustibile per produrre vapore per la generazione di energia elettrica. A tal fine, ovviamente, il potere calorifico è la più importante delle proprietà da considerare. Tuttavia, anche altre caratteristiche del carbone sono di grande importanza per la definizione delle condizioni migliori da adottare per la combustione. In generale, i carboni più vecchi si incendiano più difficilmente e richiedono camere di combustione di maggior volume per bruciare completamente. I carboni più giovani presentano una maggior reattività e possono essere bruciati velocemente in forni a ciclone. Le sostanze volatili rivestono importanza ai fini della facilità di ignizione. Le antraciti, con basso tenore di volatili, bruciano lentamente e con fiamma corta. Si prestano per quelle applicazioni per le quali il calore è trasferito direttamente dal letto di carbone. Per le combustioni in fornaci si preferiscono invece i carboni a fiamma lunga. Le percentuali di sostanze volatili e le corrispondenti frazioni del potere calorifico da esse possedute sono riportate in Tabella 3 per i diversi tipi di carboni.
Tabella 3 Sostanze volatili dei carboni fossili e loro potere calorifico.
Carbone Sostanze volatili, % Potere calorifico nelle sostanze volatili, %
Antraciti <8 5-14 Carboni magri 8-14 14-21 Carboni semigrassi 14-22 21-28 Carboni grassi corta fiamma 22-31 28-36 Carboni grassi media fiamma >31 36-47
Figura 1
Modello bidimensionale della struttura di un litantrace.

3
La tendenza al rammollimento ed alla formazione di coke non è importante quando si brucia il polverino di carbone. Nei forni a storta, invece, nei quali il carbone è bruciato in letto, è richiesto un carbone non cokificante per evitare la compattazione del letto stesso. La facilità di macinazione è naturalmente una caratteristica critica per i carboni da bruciare come polverino. Il contenuto di acqua influenza sia i costi di trasporto che la resa termica durante la combustione. I carboni più giovani, con un maggior contenuto di gruppi funzionali polari, hanno un maggior contenuto di umidità. Allorché si usano torba o lignite per la produzione di formelle combustibili, il contenuto di umidità deve essere ridotto, di solito con mezzi meccanici perché quelli termici sarebbero troppo costosi, al 15% circa. Il contenuto di ceneri e le loro caratteristiche termiche (tendenza alla fusione e formazione di scorie) rivestono una notevole importanza. Se il contenuto in ceneri è molto alto, le scorie fuse (slags) asportano una quantità notevole di calore sensibile abbassando la resa termica delle fornaci. Il contenuto in ceneri dei carboni estratti meccanicamente con apparecchiature continue può essere dell’ordine del 25% o più. Tuttavia, in genere, una riduzione di tale contenuto, ad esempio fino al 10%, mediante lavaggio, può risultare antieconomica. Le ceneri possono depositarsi sulle pareti delle fornaci dando luogo ad incrostazioni (slagging) o sulle superfici dei tubi con conseguente loro sporcamento (fouling). La tendenza allo slagging ed al fouling delle ceneri dipende dalla loro composizione chimica e, in particolare, dal loro contenuto di ossidi acidi e basici, di sodio, di calcio e magnesio, e di zolfo. Dalla composizione dipende anche un’altra importante caratteristica delle ceneri: la loro viscosità allo stato fuso. Il contenuto di zolfo riveste grande importanza in vista della qualità dei fumi e della necessità di un loro trattamento. Analoghe restrizioni circa i contenuti di ossidi di azoto e di ceneri volanti ammesse nei fumi da scaricare nell’atmosfera rendono necessaria l’adozione di sistemi di abbattimento in tutte le centrali termiche. Per quanto riguarda gli impieghi del carbone per la produzione di coke, la resa e la qualità di quest’ultimo rivestono la maggiore importanza, anche se la qualità e la quantità dei gas ottenuti come sottoprodotti sono pure considerate. I carboni migliori sono i litantraci a corta o media fiamma, col 30% circa di sostanze volatili. Le rese in coke sono del 65-70%. Gli impieghi principali dei carboni fossili consistono nella combustione (dopo eventuale desolforazione), nella carbonizzazione per l’ottenimento del coke da usare soprattutto nell’industria metallurgica, nella gassificazione e nella liquefazione. Esaminiamo brevemente nel seguito i principali processi di “conversione del carbone”, iniziando da quelli di depurazione (desolforazione) che si rendono necessari quando il carbone, per il contenuto troppo elevato di impurezze, non può essere bruciato direttamente. Pirolisi del carbone fossile
La pirolisi del carbone fossile si realizza, oggi, col fine principale di produrre il coke da utilizzare nell’industria metallurgica. Il processo di cokizzazione consiste nel riscaldamento del carbone, fuori del contatto con l’aria, a temperature introno ai 1100°C. Il coke, che rappresenta il residuo della pirolisi, è costituito quasi esclusivamente da carbonio, in varie forme cristallografiche, ma contiene anche le sostanze inorganiche inizialmente presenti nel carbone fossile, e parte dello zolfo. Durante la pirolisi, si liberano gas e vapori che rappresentano i sottoprodotti del processo. Il gas, indicato come gas di cokeria (in passato: gas illuminante), viene usato come combustibile, sia per la stessa pirolisi del carbone, sia per diversi scopi dell’industria metallurgica in vicinanza della quale l’impianto di pirolisi è collocato. La condensazione dei vapori fornisce il catrame di carbone fossile dal quale, per frazionamento e successiva raffinazione, si ricavano diversi prodotti chimici utili. I sottoprodotti più importanti ottenuti nelle cokerie sono lo zolfo, il solfato d’ammonio, gli idrocarburi aromatici, principalmente benzene e naftalina, il fenolo, la piridina, ecc. La pirolisi del carbone fossile era già impiegata commercialmente, anche se con tecnologie rudimentali, intorno al 1710, per produrre un coke che potesse utilmente rimpiazzare il carbone di

4
legna nel processo di produzione del ferro. Nei primi anni 1800, il gas di pirolisi iniziò ad essere usato come gas per illuminazione stradale. Per alcuni impianti il gas divenne il prodotto principale, mentre il residuo della pirolisi rappresentava un fastidioso sottoprodotto. Nella seconda metà del 19° secolo, inoltre, in Germania cominciò uno sfruttamento sempre crescente dei prodotti liquidi (catrame) dai quali potevano ottenersi le materie prime che consentirono lo sviluppo di alcune importanti produzioni chimiche. Lo sviluppo dell’industria chimica che, iniziato negli ultimi anni del secolo diciannovesimo culminò nei primi decenni del ventesimo, avvenne grazie all’uso sempre più intensivo dei sottoprodotti della pirolisi del carbone. In effetti, l’industria chimica sviluppatasi in quegli anni, e fino al 1940 circa, è comunemente indicata come “carbochimica”, così come quella sviluppatasi successivamente, e basata sull’uso prevalente del petrolio come fonte di materie prime, è comunemente indicata come “petrolchimica”. Lo sviluppo crescente dei processi di pirolisi portò ad una disponibilità crescente di coke, alla quale si accompagnò uno straordinario sviluppo dei processi metallurgici e, in particolare, siderurgici. La produzione mondiale di coke metallurgico è valutata oggi attorno alle 380⋅106 t/anno, con un tasso di crescita globale molto modesto (<0,3% annuo) a causa della maturità dell’industria siderurgica nel mondo occidentale. Nei paesi occidentali ed alleati (America, Europa occ.le, Giappone, Africa e Medio Oriente, Asia), infatti, la produzione complessiva è di circa 190⋅106 t/anno, con un tasso di crescita pari a –1% l’anno, mentre nei paesi dell’est (Russia, Cina ed Europa or.le) si ha la stessa produzione di 190⋅106 t/anno, ed un tasso annuo di crescita del ∼2%. Le riserve di carbone fossile ancora disponibili variano, secondo le stime effettuate nel 1990, tra i 1000 e i 1600 miliardi di tonnellate. Solo 1/10 circa del carbone fossile disponibile è adatto per la produzione di coke metallurgico. Considerando le rese di pirolisi, si stima dunque che il coke ottenibile dalle riserve sia sufficiente per coprire le necessità dell’industria metallurgica per altri 200 anni, con i consumi attuali. Per la produzione di coke metallurgico, il carbone fossile deve presentare una serie di caratteristiche correlate, prevalentemente, al tenore di sostanze volatili ed alla tendenza al rammollimento per riscaldamento. Infatti, un buon coke metallurgico, oltre ad assolvere le funzioni di combustibile (per generare il calore necessario per raggiungere le temperature richieste per la riduzione dei minerali) e di reagente (per produrre i gas riducenti o agire esso stesso come riducente per gli ossidi metallici), deve anche presentare una notevole resistenza meccanica, per non sgretolarsi durante le operazioni di trasporto e di caricamento dei forni e, soprattutto, per resistere alle forti pressioni generate dal peso degli strati superiori di materiale, nei forni stessi, e deve infine conservare la porosità sufficiente per consentire il passaggio dei gas verso l’alto e quello del metallo fuso verso il basso. Tutte queste caratteristiche possono essere raggiunte nel modo migliore con una ottimizzazione opportuna della qualità della carica delle fornaci di pirolisi. È infatti opportuno che il carbone possa rammollire, durante le fasi intermedie del riscaldamento, in modo da dare una massa pastosa che, per ulteriore pirolisi, si trasforma infine in un materiale solido poroso (per i vuoti lasciati dal passaggio dei prodotti gassosi) ma con struttura piuttosto compatta a livello microscopico. Come già precedentemente accennato, i carboni che si prestano meglio per la produzione di coke sono i litantraci a corta o media fiamma, col 30% circa di sostanze volatili. Nella pratica industriale, si usa sempre miscelare carboni di provenienza diversa, sia per ragioni commerciali che per ottimizzare la qualità della carica alle fornaci di cokificazione. Le cokerie sono in generale situate nelle vicinanze degli impianti metallurgici. Il carbone fossile deve quindi essere trasportato dalle zone di estrazione mediante navi, ferrovia, autotreni ecc.

5
Lo schema di un impianto di cokificazione e di trattamento dei prodotti è mostrato in Figura 2.
Figura 2
Schema di un impianto di cokificazione. Ciascun tipo di carbone viene stoccato separatamente. Si procede quindi alla miscelazione
ed alla frantumazione e macinazione della miscela. Con la frantumazione si riduce la pezzatura del carbone a meno di circa 2 cm rimuovendo le pietre e gli altri materiali estranei, in particolare metallici. La macinazione viene fatta in mulini a martelli ed è spinta fino a raggiungere una granulometria del carbone con almeno l’80% delle particelle di dimensioni inferiori ai 3 mm. Uno schema di moderna fornace a coke è rappresentato nella Figura 8. L’impianto, costruito in mattoni refrattari, consta di una batteria di circa 80 fornaci separate l’una dall’altra da intercapedini attraverso le quali passano i fumi caldi della combustione. Nella parte sottostante la batteria di fornaci sono disposti i rigeneratori di calore e le condotte per il preriscaldamento dell’aria e del gas combustibile, pure costruiti in mattoni refrattari. Le singole fornaci hanno una lunghezza (equivalente alla larghezza della batteria) di 12-14 m ed una altezza interna di 4-6 m. La larghezza interna delle fornaci è di circa 40 cm ad una estremità e circa 50 cm all’altra. La sezione orizzontale è dunque trapezoidale, e ciò serve a rendere agevole lo svuotamento delle fornaci alla fine della pirolisi. Lo svuotamento viene effettuato aprendo le due porte poste alle estremità laterali delle singole fornaci e spingendo il coke, mediante un pistone, ad uscire dalla porta di larghezza maggiore ed a cadere su un carrello che scorre su binari disposti parallelamente alla batteria. Le porte sono costruite in acciaio rivestito internamente con refrattario. Ciascuna singola fornace può contenere circa 20 t di carbone.

6
Figura 3
Schema di batteria di forni di cokificazione. A sinistra: sezione longitudinale (parziale); a destra doppia sezione trasversale: attraverso una fornace (a destra) e attraverso le condotte di riscaldamento (a sinistra).
Il caricamento delle fornaci con la polvere di carbone viene fatto dall’alto, attraverso 3-5 fori di alimentazione, facendo cadere la polvere trasportata da carrelli che scorrono su binari disposti longitudinalmente, sopra la batteria. I carrelli sono a loro volta caricati con la polvere di carbone contenuta negli appositi sili di stoccaggio disposti ad una estremità della batteria. Al momento del caricamento, che avviene immediatamente dopo lo svuotamento, le fornaci sono ancora molto calde. Infatti, una volta riscaldata, la batteria deve essere mantenuta a temperatura elevata per tutta la sua vita, per evitare gli inevitabili danni che sarebbero altrimenti causati dalle forti contrazioni e dilatazioni termiche e dalle variazioni mineralogiche irreversibili subite dai refrattari silicei. Al termine del caricamento delle fornaci, e dopo la chiusura dei relativi fori, si procede, mediante apertura di un altro foro praticato nella parte alta di una delle porte laterali, dal quale passa una barra metallica che viene mossa orizzontalmente avanti e indietro, alla spianatura della parte superiore della carica, in modo da lasciare uno spazio uniforme tra questa e la parete superiore della fornace. Ciò consente di avere un riscaldamento più uniforme e permette ai gas sviluppati dalla pirolisi di scorrere facilmente verso i fori di uscita, situati ad una o ad ambedue le estremità laterali delle pareti superiori delle fornaci. I fori sono collegati a tubi verticali rivestiti internamente di refrattario che si connettono, mediante un collo d’oca che garantisce la tenuta idraulica, ad un canale collettore che corre lungo la batteria. In questo canale, alimentato continuamente con acqua ammoniacale fredda, si realizza un primo raffreddamento, con parziale condensazione della miscela di gas e vapori che escono dalle fornaci. Nella maggior parte degli impianti viene caricato carbone umido. Ciò comporta che già all’atto del caricamento delle fornaci e nelle fasi iniziali del riscaldamento si ha un forte sviluppo di vapore d’acqua. In alcuni impianti si realizza invece un preriscaldamento del carbone a 150-200°C

7
e ciò consente di accorciare i cicli di pirolisi. Questi hanno una durata variabile tra le 16 e le 24 ore. Le operazioni di caricamento e di svuotamento delle fornaci sono programmate in modo da ottimizzare tutti gli effetti con esse connessi e, in particolare, le pressioni generate sulle pareti di fornaci adiacenti, le quantità di carbone che devono restare sulle pareti interne delle fornaci dopo lo svuotamento, ecc. Dato che le pareti sono costruite con mattoni refrattari, praticamente senza malta, la tenuta delle fornaci è affidata al carbone depositato a seguito della pirolisi, il cui spessore deve essere tuttavia non troppo elevato per non limitare il volume utile delle camere. Al momento dello scarico del coke rovente, sia questo che il carbone che resta sulle pareti delle fornaci tende ad incendiarsi. La combustione del carbone depositato sulle pareti viene interrotta chiudendo nuovamente le porte del forno e lasciando uscire l’anidride carbonica attraverso appositi fori. Il coke rovente viene rapidamente trasportato dal carrello, pure rivestito di materiale refrattario, sotto un serbatoio dal quale viene spruzzata una quantità di acqua appena sufficiente allo spegnimento, in modo che il coke rimanga asciutto. Il processo di cokificazione si realizza attraverso un progressivo riscaldamento della carica fino a temperature di 1000-1200°C, condotto in modo da escludere la presenza di ossigeno. Il riscaldamento provoca una serie di reazioni che portano alla evoluzione di prodotti leggeri, relativamente ricchi di idrogeno, con progressivo arricchimento del residuo in carbonio, ed alla sinterizzazione spinta delle particelle di carbone, attraverso la loro transitoria plasticizzazione, con formazione di una massa di coke solida e porosa. Al momento del caricamento della polvere di carbone nella fornace calda, le particelle di carbone in contatto con la parete fondono rapidamente, saldandosi le une alle altre, ed iniziano a devolatilizzare. La perdita progressiva di prodotti leggeri ricchi di idrogeno comporta una graduale nuova solidificazione di questo strato plastico di carbone, mentre il calore si propaga verso le zone più interne della massa di carbone e il guscio di materiale che si trova nello stato plastico e che subisce sinterizzazione si muove gradualmente verso l’interno. Il processo è schematizzato nella Figura 9, nella quale il guscio di materiale plastico che separa la zona più esterna, costituita da coke e semicoke, e quella interna costituita da carbone in via di plasticizzazione è rappresentato in nero. Questo strato di materiale semifuso rappresenta una barriera al passaggio dei gas. Il gas generato entro lo strato fuso può migrare sia verso l’esterno che verso l’interno; ma quello che si forma fuori o dentro il guscio difficilmente riesce ad attraversarlo. Nella parte più calda del guscio, verso le pareti della fornace, la devolatilizzazione trasforma il materiale fuso in un solido poroso indicato come “semicoke”. Questo contiene ancora quantità rilevanti di materiale pirolizzabile che subisce una ulteriore devolatilizzazione man mano che altro calore è trasferito dalle pareti della fornace e la temperatura si avvicina ai 1000°C. Queste trasformazioni chimiche che avvengono nello stato solido trasformano il semicoke in coke. Contemporaneamente si ha anche una trasformazione fisica del materiale con aumento del grado di cristallinità e della orientazione della struttura e con riduzione parziale della porosità ed aumento della resistenza meccanica. All’interno del guscio fuso, la cui temperatura media è, indicativamente, nell’intervallo di 250-400°C, la temperatura resta per molte ore intorno ai 100°C, fino a
Figura 4
Sviluppo e migrazione dello strato plastico che separa il coke (esterno) dal carbone (interno). Situazione a metà (a), a circa 2/3 (b) ed alla fine (c) del ciclo di pirolisi.

8
quando tutta l’acqua non è stata allontanata come vapore. Quando il guscio di materiale fuso ha raggiunto il centro della fornace (schema b, in Figura 9) la temperatura varia, attraverso la massa di semicoke, dai 1200°C circa delle pareti, ai 400°C circa della zona più interna. Col progredire del riscaldamento, il gradiente di temperatura tende a diminuire, finché, alla fine del ciclo di pirolisi (mediamente di 15-20 ore) la temperatura interna ha raggiunto i 980°C circa. La progressiva devolatilizzazione del semicoke e la sua graduale trasformazione in coke comporta di solito un certo ritiro, dovuto alla riorganizzazione degli atomi di carbonio in una struttura cristallina più compatta. Questo effetto, di per se, tenderebbe a far contrarre la massa di carbone ed a provocarne il distacco dalle pareti della fornace. Tuttavia, altri effetti come lo sviluppo di sostanze gassose all’interno del guscio fuso, con conseguente aumento della pressione, e la dilatazione termica della carica in riscaldamento, contrastano questa tendenza alla contrazione di volume e fanno in modo che la carica continui ad esercitare una certa pressione sulle pareti della fornace. Ciò favorisce la trasmissione del calore dalle pareti alla carica. Tuttavia se la pressione dovesse salire oltre certi limiti si creerebbero situazioni di rischio per la stabilità delle pareti le quali potrebbero anche essere danneggiate per il forte attrito, al momento dello svuotamento dei forni. Risulta quindi molto importante determinare le caratteristiche della carica, attraverso una progettazione accurata della miscela di carbone di alimentazione, nonché le condizioni del processo in modo da avere un andamento ottimale delle contrazioni della carica e delle pressioni esercitate sulle pareti. Nello schema di Figura 10 sono riportati gli andamenti tipici dei due più importanti parametri di processo durante un ipotetico ciclo di pirolisi di 18 ore. La miscela di gas e vapori liberata durante la pirolisi del carbone lascia le fornaci attraverso i tubi appositi ed entra nel canale collettore ove viene continuamente spruzzata l’acqua ammoniacale di riciclo, fredda. Nel collettore si ha la condensazione del catrame e di parte del vapor d’acqua e la dissoluzione dell’ammoniaca. La miscela bifasica liquida (acqua/catrame) scorre nel collettore, assieme ai gas, per raccogliersi in un serbatoio di decantazione. I gas passano ad uno scambiatore dove sono raffreddati con acqua a 30°C. Essi sono quindi compressi, raffreddati nuovamente ed inviati ad un estrattore a labirinto nel quale si separano le ultime goccioline di catrame trascinate. Il catrame proveniente dal decantatore e dall’estrattore è inviato all’impianto di frazionamento.
L’acqua ammoniacale, con l’1% circa di ammoniaca, viene in parte raffreddata e riciclata al tubo collettore e, per la parte rimanente, inviata ad un estrattore nel quale, per aggiunta di latte di calce ed iniezione di vapore, si separa l’ammoniaca dal liquido che viene scaricato. I vapori ammoniacali, assieme ai gas non condensati nel separatore sono inviati ad un saturatore nel quale si trova una soluzione di acido solforico al 5-10%, mantenuta a 60°C. Qui si forma solfato ammonico cristallino che viene separato dal basso come una torbida che è inviata ad una centrifuga per la separazione delle acque madri da riciclare. La concentrazione dell’acido nel saturatore è mantenuta costante per aggiunta di acido concentrato.
I gas, ulteriormente raffreddati, sono inviati ad una torre di lavaggio dove è spruzzato dall’alto un olio pesante ottenuto dal frazionamento del petrolio. I vapori di idrocarburi (principalmente benzene) ancora presenti nel gas sono così separati e recuperati.
Figura 10 Andamento della temperatura interna e della pressione.
T, °C 1000
800
600
400
200
4 8 12 16 t, ore
P, kPa
24
18
12
6

9
Il gas residuo è bruciato direttamente, per il 40% circa, per fornire il calore necessario alla pirolisi. Il rimanente viene prevalentemente usato come combustibile negli impianti metallurgici adiacenti. La composizione media del gas è la seguente:
H2 50%; CH4+ 36%; CO 8%; CO2 3%; N2 3%
Il catrame di carbone fossile, preriscaldato a 120-150°, viene riscaldato con pece di riciclo fino a circa 350°C ed inviato ad una colonna di frazionamento dalla quale si ottengono diverse frazioni:
1. L’olio leggero (∼5%), costituito prevalentemente da idrocarburi aromatici (benzene, toluene, xileni) e piridina, con punto di ebollizione fino a 170°C;
2. L’olio “carbolico” (∼17%), contenente fenolo ed altri composti aromatici, con intervallo di ebollizione 170-205°C;
3. L’olio di naftalina (∼7%), dal quale si estrae la naftalina, purificata poi per sublimazione, con intervallo di ebollizione 205-240°C;
4. L’olio di “creosoto” (∼4%), contenente numerosi idrocarburi aromatici a nuclei condensati, con intervallo di ebollizione 240-280°C;
5. L’olio di antracene (∼5%), costituito da una miscela di composti, prevalentemente aromatici con tre o più anelli condensati, con intervallo di ebollizione 270-340°C;
6. Il residuo, o pece, (∼62%), con punto di ebollizione superiore a 325°C. Di queste frazioni, le uniche che rivestono ancora oggi un discreto valore commerciale sono
l’olio leggero, dal quale, in passato, si estraevano gli idrocarburi aromatici da impiegare come materie prime per l’industria chimica e tutta la piridina disponibile sul mercato, l’olio di naftalina, dal quale si ottiene ancora oggi una discreta frazione della naftalina necessaria, ed il residuo che viene usato come bitume impermeabilizzante.
Le rese indicative di prodotti ottenibili per pirolisi di 1 t di carbone fossile sono: � Coke 715 Kg
� Polverino di coke 46 Kg
� Catrame 39 Kg
� Solfato d’ammonio 10 Kg
� Olio leggero (dal lavaggio dei gas) 10 Kg
� Gas 18 Kg
Gassificazione del carbone
La gassificazione del carbone, già impiegata fin dal 1800 per produrre un gas combustibile da impiegare per illuminazione e riscaldamento civile, ha avuto un notevole sviluppo, in molti paesi industrializzati, fino al 1940, allorché si è avuta una larga disponibilità di gas naturale a basso costo.
Storicamente, il primo processo di gassificazione del carbone era realizzato per reazione del carbone con aria in difetto e portava al così detto gas d’aria. Questo si otteneva facendo passare una miscela di aria e vapore d’acqua attraverso un letto di un combustibile carbonioso solido (carbone fossile o coke) incandescente. La temperatura del letto veniva scelta in funzione della temperatura di fusione delle ceneri ed era regolata aggiustando il rapporto tra l’aria e l’acqua nella miscela gassosa. Essa variava, normalmente, tra 1000 e 1500°C.
Il vapor d’acqua ha lo scopo di usare il calore prodotto dalla reazione del carbone con l’aria per reagire endotermicamente col carbone stesso e fornire una miscela di ossido di carbonio e idrogeno. Il processo risulta complessivamente autotermico. La temperatura del letto e la percentuale di vapore d’acqua nella miscela gassosa di alimentazione possono essere aumentate recuperando al meglio il calore sensibile dei gas prodotti per riscaldare quelli alimentati. Le reazioni coinvolte nel processo sono:
C + O2 + 4 N2 (aria) → CO2 + 4 N2 ∆H1000°C = -94,6 Kcal/mole (1)(1)(1)(1)

10
CO2 + C → 2 CO ∆H1000°C = +40,2 Kcal/mole (2)(2)(2)(2)
C + H2O → CO + H2 ∆H1000°C = +32,5 Kcal/mole (3)(3)(3)(3)
CO + H2O ↔ CO2 + H2 ∆H1000°C = -7,7 Kcal/mole (4)(4)(4)(4)
L’altezza del letto di carbone è consistente (0,6 – 1,8 m). L’aria reagisce dapprima secondo la reazione (1) per dare anidride carbonica. Negli strati successivi di carbone, quest’ultima si riduce ad ossido di carbonio. Contemporaneamente, l’acqua reagisce col carbone per dare ossido di carbonio e idrogeno, mentre nella fase gassosa, si stabilisce l’equilibrio della reazione (4). Il gas d’aria ha un potere calorifico molto basso (meno di 1/7 di quello del gas naturale) ed il suo impiego è ormai completamente abbandonato. Un miglioramento tecnologico introdotto successivamente consentì di ottenere un combustibile gassoso con potere calorifico migliorato: il gas d’acqua. Il gas d’acqua (indicato dagli inglesi come blue gas per il colore della fiamma) viene ottenuto per passaggio di vapore d’acqua attraverso un letto di carbone incandescente, secondo la reazione (3). Dato che la reazione è endotermica, la temperatura del letto di carbone si abbassa da un valore iniziale di 1000-1100°C fino a circa 900°C, dopo un tempo di 5-6 minuti. Questa fase della reazione, indicata come “soffio
freddo”, deve quindi essere interrotta per scaldare nuovamente il letto di carbone mediante immissione di aria (“soffio caldo”). Il soffio caldo ha una durata di circa 2 min e riporta la temperatura del letto a 1000-1100°C. Nella fase iniziale del soffio caldo, quando la temperatura del letto è più bassa, i fumi contengono una percentuale notevole di anidride carbonica, prodotta dalla reazione (1). Successivamente, l’ossidazione avviene secondo la reazione (5), somma delle reazioni (1) e (2), che è molto meno esotermica.
2 C + O2 + 4 N2 (aria) → 2 CO + 4 N2 ∆H1000°C = -54,4 Kcal/mole (5)(5)(5)(5)
Il processo si realizza attraverso una successione di soffi caldi e freddi ed il gas d’acqua prodotto è quello ottenuto come effluente dei soffi freddi, mentre i gas prodotti dal soffio caldo sono separati come fumi. La tecnica permette di eliminare il più possibile l’azoto dell’aria che abbassa il potere calorifico della miscela, anche se un certo miscelamento dei gas prodotti nel soffio caldo e nel soffio freddo avviene inevitabilmente all’atto dello scambio. Il gas d’acqua ha un potere calorifico quasi doppio di quello del gas d’aria; esso è tuttavia ancora piuttosto basso per le applicazioni pratiche. Allo scopo di aumentare il potere calorifico del gas d’acqua, è possibile spruzzare dell’olio (residui di basso pregio dell’industria petrolifera) nei gas caldi che escono dalla fornace, provocandone così una pirolisi spinta che arricchisce il gas di metano e di idrocarburi illuminanti. Si ottiene in questo modo il gas d’acqua carburato, con potere calorifico quasi raddoppiato.
In epoche più recenti, si sono avuti notevoli sviluppi nella tecnologia di gassificazione del carbone, in particolare attraverso l’uso di ossigeno, in luogo dell’aria, e di alte temperature ed alte pressioni. In particolare, la disponibilità di ossigeno a basso prezzo, conseguente alla commercializzazione su vasta scala dei processi di distillazione dell’aria per la produzione di azoto da impiegare come materia prima per la sintesi dell’ammoniaca, ha consentito di sfruttare i vantaggi del processo di produzione del gas d’aria (processo continuo, realizzato in condizioni di reazione stazionarie) con quelli del processo di produzione del gas d’acqua (assenza di azoto nel prodotto). I moderni processi di gassificazione del carbone, consistono nel far passare una miscela opportunamente dosata di vapore d’acqua ed ossigeno attraverso un letto di carbone. In realtà, qualunque sostanza carboniosa può essere gassificata con questo metodo, ed il gas prodotto può essere utilizzato oltreché come combustibile, anche come gas di sintesi (si indicano con questo nome le miscele di idrogeno ed ossido di carbonio, con rapporto tra i due componenti variabile, che sono usati per processi di sintesi). Come descritto in seguito, in molti paesi che dispongono di rilevanti quantità di gas naturale, il gas di sintesi viene prodotto utilizzando metano come materia

11
prima. In altri paesi, come il Giappone, nei quali la disponibilità di gas naturale è ridotta, si può produrre gas di sintesi a partire da nafte. Processi di gassificazione analoghi a quelli realizzati a partire da carbone possono poi essere realizzati utilizzando biomasse, residui agricoli (ad esempio i residui solidi della estrazione dello zucchero dalla canna) e, addirittura, rifiuti solidi urbani.
Le reazioni coinvolte nei processi di gassificazione sono sostanzialmente identiche a quelle già precedentemente considerate e che si riportano sotto per comodità. Accanto a queste reazioni, se ne devono considerare altre, come quelle di metanazione (6), di ossidazione dell’idrogeno (7), ecc., che complicano notevolmente il quadro.
C + O2 → CO2 ∆H1000°C = -94,6 Kcal/mole (1)(1)(1)(1)
CO2 + C ↔ 2 CO ∆H1000°C = +40,2 Kcal/mole (2)(2)(2)(2)
2 C + O2 → 2 CO ∆H1000°C = -54,4 Kcal/mole (5)(5)(5)(5)
C + H2O → CO + H2 ∆H1000°C = +32,5 Kcal/mole (3)(3)(3)(3)
CO + H2O ↔ CO2 + H2 ∆H1000°C = -7,7 Kcal/mole (4)(4)(4)(4)
CO + 3 H2 ↔ CH4 + H2O ∆H1000°C = -52,2 Kcal/mole (6)(6)(6)(6)
H2 + ½ O2 → H2O ∆H1000°C = -57,8 Kcal/mole (7)(7)(7)(7)
Dato che molte delle reazioni soprascritte sono di equilibrio, esse saranno spostate più o
meno verso destra a seconda della temperatura e della pressione. In particolare, un aumento di pressione da 1 a 40 atmosfere comporta un aumento del contenuto di metano dei gas da valori trascurabili fino ad oltre il 30%, con contemporanea riduzione del contenuto di ossido di carbonio e di idrogeno e con sensibile aumento del potere calorifico da meno di 3000 ad oltre 4700 Kcal/m3. In alcuni reattori di gassificazione (idrogassificatori) si può inviare col carbone anche un gas ricco di idrogeno, con lo scopo di incrementare il contenuto di metano nei gas ed aumentare ulteriormente il potere calorifico.
Negli ultimi due decenni, anche in seguito alla crisi petrolifera del 1973 ed ai conseguenti aumenti del prezzo del petrolio e del gas naturale, la gassificazione del carbone è stata di nuovo considerata dalle aziende produttrici di energia elettrica come una delle tecniche di maggior interesse per il futuro. Malgrado le iniziative intraprese da molti stati per limitare l’uso dell’energia elettrica, si prevede che, almeno nei paesi più sviluppati come gli Stati Uniti, la richiesta di questo tipo di energia crescerà, nel decennio 2000-2010, con un gradiente circa doppio di quello della richiesta complessiva di energia. D’altra parte, la combustione diretta del carbone fossile pone notevoli problemi tecnologici legati soprattutto alla necessità di contenere entro limiti accettabili l’impatto ambientale, come descritto in precedenza. Per tutto quanto sopra, la moderna tecnica di produzione di energia elettrica a ciclo combinato, con l’uso di gas prodotto per gassificazione del carbone si offre come una alternativa di rilevante interesse. Il ciclo combinato con gassificazione del carbone (coal gasification combined cycle - CGCC) realizza due tecnologie vincenti: la prima consiste nella produzione di un gas a combustione pulita a partire da un combustibile fossile di larga disponibilità quale il carbone; la seconda consiste nell’uso di tale gas per produrre energia elettrica con ciclo combinato ad alta efficienza. Quest’ultimo consiste in un primo stadio di combustione in turbine ad alta temperatura, con produzione diretta di energia elettrica, ed in un secondo stadio nel quale si recupera il calore dei fumi esausti per produrre vapore che aziona una seconda batteria di turbine. Uno schema del processo è mostrato nella Figura 11.

12
Carbone Gasificazione Depurazionegas
Turbinea gas
OssigenoRecupero
calore
Scorie Zolfo
Turbinea vapore
Sottoprodotti commerciabili
Elettricità
Figura 11
Schema di un processo di produzione di energia elettrica con la tecnologia CGCC.
Il sistema CGCC garantisce un resa energetica del 42% circa, calcolata come rapporto tra l’energia elettrica generata ed il potere calorifico superiore del carbone, contro le rese del 35% circa raggiungibili nei comuni impianti di combustione del carbone con generazione di vapore. I processi di gassificazione consistono essenzialmente nel far reagire il carbone con miscele di vapor d’acqua ed aria o, più comunemente, di vapor d’acqua ed ossigeno. La reazione con l’ossigeno produce il calore necessario per mantenere una temperatura sufficientemente alta da consentire una cinetica soddisfacente e per compensare il calore assorbito dalla reazione col vapore. La chimica dei processi di gassificazione segue le reazioni (1) – (7) riportate sopra. Gli impianti di gassificazione del carbone più usati oggi sono essenzialmente di tre tipi: gassificatori a letto mobile (reattori in controcorrente), gassificatori a letto fluido (reattori miscelati); gassificatori a letto trascinato (reattori con flusso a pistone). Essi sono schematizzati in Figura 12.
Figura 12 Tipi di gassificatori: (a) letto mobile; (b) letto fluido; (c) letto trascinato.
Nei gassificatori a letto mobile, esemplificati dal classico gassificatore Lurgi, il carbone con pezzatura da 5 a 50 mm viene alimentato dall’alto e si scalda progressivamente a spese del calore sensibile dei gas che salgono. Questi escono dal reattore a 300°C, se si usano ligniti umide, o a
13
550°C, se si usano litantraci. Man mano che il carbone scende verso il basso, nel letto, si scalda progressivamente subendo dapprima la devolatilizzazione e poi la cokificazione. Ancora più in basso, esso reagisce con il vapor d’acqua e con l’ossigeno per dar luogo alla gassificazione vera e propria. Il letto di carbone scende gradualmente verso il basso man mano che il solido si consuma e le ceneri attraversano la griglia di sostegno del letto per uscire dal basso, dopo aver ceduto parte del calore sensibile per scaldare la miscela ossigeno/vapore. Caratteristica saliente dei gassificatori a letto mobile è la richiesta relativamente bassa di ossigeno resa possibile dal recupero termico spinto. Essa comporta una temperatura di gassificazione modesta ed una elevata percentuale di metano nei gas, che sono anche accompagnati da idrocarburi liquidi come oli e catrame. Le basse temperature nella zona di combustione non permettono la scorificazione delle ceneri. In questi processi risulta difficile lavorare con carbone di granulometria troppo fine ed anche con carboni con tendenza troppo spinta al rammollimento. Nei gassificatori a letto fluido la temperatura è uniforme e moderata (normalmente, al di sotto di quella di scorificazione delle ceneri). Il letto è costituito dalle particelle di carbone, che sta subendo la devolatilizzazione, e di char che reagisce col vapore e l’ossigeno. Le particelle ormai fortemente assottigliate di char possono essere trascinate fuori dal reattore, assieme alle ceneri volanti, dai gas. Esse sono recuperate riciclando le ceneri volanti, separate in cicloni, alla parte più bassa del letto fluido. In alcuni gassificatori, che lavorano a temperatura leggermente maggiore, si ha la agglomerazione parziale delle ceneri nella zona inferiore, più calda, del letto fluido. Nei gassificatori a letto trascinato, il polverino di carbone reagisce in equicorrente con la miscela ossigeno/vapore salendo attraverso il reattore con tempi di residenza di alcuni secondi. Questi sistemi lavorano ad alte temperature e consentono la conversione completa del carbone e l’eliminazione delle ceneri scorificate dal basso. Essi possono usare qualunque tipo di carbone e producono gas quasi privi di frazioni liquide, facilitandone così anche la successiva depurazione. I più importanti parametri operativi dei gassificatori sono: la composizione del carbone di partenza (lignite, litantrace, antracite, coke di petrolio), il rapporto ossigeno/carbone ed il rapporto ossigeno/vapore. Questi ultimi due parametri sono funzioni del primo: i carboni più giovani (ligniti) richiedono meno ossigeno ed una quantità di vapore molto piccola o addirittura nulla, dato che hanno un contenuto relativamente alto di ossigeno ed idrogeno; quelli più vecchi (antraciti) sono meno reattivi e richiedono più ossigeno e quantità più alte di vapore. L’efficienza di un processo di gassificazione è valutata attraverso la resa in gas, la sua composizione, la conversione del carbone ed il valore energetico del gas freddo (CGE, cold gas efficiency), espresso come il rapporto tra il contenuto energetico del gas e quello del carbone di partenza. I valori tipici dei suddetti parametri sono riportati in Tabella 4.
Tabella 4 Esempi di processi di gassificazione di carboni.
Parametro Lignite Litantrace Coke di
petrolio
Rapporto ossigeno/carbonio Rapporto vapore/ossigeno Resa in gas (Kg/Kg carbone) Composizione gas (vol %) CO H2 CO2 H2S + COS N2 + CH4 Conversione del carbone (%)
0,87 -
0,95
61 28 5
0,8 5,2
99,6
0,97 0,15 1,6
64 30 2
0,4 3,6
99,7
1,04 0,23 2,1
65 26 2 2 5
99,5

14
CGE (%) 79 83 80
Come può notarsi, la conversione del carbone è elevatissima per tutti i processi moderni, indipendentemente dal tipo di carbone alimentato. Anche la composizione del gas varia poco al variare del tipo di carbone; il suo potere calorifico superiore, dopo rimozione dei composti solforati (H2S e COS) e della CO2, è di circa 2900 Kcal/m3. La resa ponderale in gas è vicina all’unità per le ligniti, mentre si avvicina a 2 per le antraciti ed il coke di petrolio. Il parametro CGE, espresso come detto sopra dal rapporto tra l’energia chimica del gas e quella del carbone di partenza è attorno all'80%. Tale parametro è calcolato sulla base del gas freddo. Il bilancio energetico complessivo dei processi di gassificazione, che deve tenere conto anche del calore recuperato per raffreddamento del gas, ecc., è normalmente vicino al 95%. Il gas ottenuto per gassificazione del carbone può essere impiegato, oltreché per la combustione e la produzione di energia, come esemplificato per i processi CGCC (V. p. 16), anche per la sintesi di ammoniaca, dopo conversione del CO ad H2, per la sintesi del metanolo, dopo conversione fino ad ottenere un rapporto molare H2/CO pari a 2, per le sintesi di idrocarburi (benzine, paraffine, alcoli) con i processi di Fischer e Tropsch, per la sintesi di aldeidi ed alcoli con il processo OXO, dopo conversione fino ad ottenere un rapporto molare H2/CO pari ad 1, ecc. Per questo motivo, come già accennato in precedenza, il gas ottenuto per gassificazione del carbone viene anche designato come gas di sintesi, analogamente a quello che può essere prodotto a partire da gas naturale, come descritto nel seguito. I processi di produzione di sostanze organiche liquide di diverso tipo a partire da carbone, attraverso la gassificazione di quest’ultimo e la successiva reazione di CO con H2, con proporzioni molari diverse, vanno sotto il nome di processi di liquefazione indiretta del carbone.
15
Capitolo 2
IL GAS NATURALE Gas naturale
A partire dal 1950, si è avuto uno sviluppo straordinario delle tecniche di estrazione, di trasporto e di utilizzo del gas naturale, che ha portato ad una vera rivoluzione del mercato dei gas combustibili. La disponibilità di quantità notevoli di gas naturale e di idrocarburi ricavati da petrolio ha limitato fortemente, negli ultimi cinquanta anni, l’importanza dei processi di produzione di gas combustibili da carbone fossile. Tuttavia, la contrazione delle disponibilità future di gas naturale e di petrolio ed il loro conseguente aumento di prezzo, lasciano prevedere un nuovo capovolgimento della situazione a favore del carbone fossile, anche se questo si verificherà, molto probabilmente, in modo graduale. Di conseguenza, come descritto nel precedente capitolo, negli ultimi anni sono stati studiati e, in alcuni paesi (ad es. il Sud Africa), realizzati processi migliorati per la produzione di gas combustibili puliti e con potere calorifico elevato, a partire da carbone. In alcuni casi, per indisponibilità di gas naturale, si è addirittura prodotto metano da carbone o da residui petroliferi attraverso processi di “metanazione”. Il metano così prodotto è comunemente indicato con la sigla SNG (synthetic natural gas). Il gas naturale ha origini e meccanismi di formazione simili a quelli del petrolio e si trova in giacimenti che possono essere in prossimità di quelli del grezzo, oppure lontani da essi. L’estrazione del petrolio comporta spesso l’ottenimento di “gas associato” che deve essere separato dal grezzo liquido, prima che questo possa essere introdotto nei serbatoi di trasporto (petroliere) o negli oleodotti. Questo gas associato è ricco di idrocarburi “illuminanti”, cioè degli idrocarburi aeriformi (gas e vapori) con numero d’atomi di carbonio fino a 4 (Tabella 1).
Tabella 1
Costanti critiche e tensioni di vapore degli idrocarburi paraffinici
Idrocarburo Tc
(°C) Pc
(atm) Teb (°C)
Pvap(25°C) (atm)
Metano Etano Propano n-Butano n-Pentano
-82,5 32,1 96,8
153,0 197,2
45,8 48,8 42,0 36,0 33,0
-161,5 -88,6 -42,1 -0,5 36,2
- 42 10 2,2 0,7
I gas naturali vengono detti “secchi” quando il contenuto di idrocarburi illuminanti è sufficientemente basso da non consentirne la condensazione per compressione (fino ai valori normalmente usati nelle operazioni di depurazione, trasporto e stoccaggio) e per raffreddamento (fino ai valori normalmente raggiungibili durante il trasporto in gasdotti). Quelli con contenuti superiori di idrocarburi illuminanti, ivi compresi i “gas associati” (al petrolio), sono detti “umidi”. Il potere calorifico dei gas naturali è nettamente maggiore di quello dei gas prodotti da carbone, come mostrato nella Tabella 2 nella quale sono dati i poteri calorifici di alcuni composti puri nonché le composizioni ed i poteri calorifici dei più comuni gas combustibili industriali. Tra questi, quelli prodotti a partire da carbone, già trattati nel precedente capitolo, sono indicati in corsivo. Per questi, così come per i due tipi di gas naturale indicati (secco e umido) viene dato il contenuto cumulativo di idrocarburi illuminanti (C2-C5).
Il GPL (gas di petrolio liquefatto) (in inglese: LPG = liquified petroleum gas o low pressure
gas) si ottiene dal prodotto di testa della distillazione primaria del petrolio o dalla stabilizzazione dei gas naturali umidi.
Si nota immediatamente, dai dati della Tabella 2, che il potere calorifico degli idrocarburi gassosi, che è generalmente misurato per ovvi motivi con riferimento all’unità di volume, cioè con riferimento ad uguali numeri di moli, cresce sensibilmente al crescere della massa molecolare. Per questa ragione, nei bruciatori, è necessario regolare in modo opportuno il rapporto aria/combustibile in modo da avere quello più adatto alla combustione dei diversi gas (ad esempio, metano o GPL).
16
Tabella 2
Composizioni e poteri calorifici di gas combustibili.
Gas
Composizione Potere
calorifico
CO CO2 H2 N2 O2 CH4 C2H6 C3H8 C4H10 C5H12 (Mcal/m3)
Idrogeno - - 100 - - - - - - - 2,580
Ossido di carbonio 100 - - - - - - - - - 3,019
Metano - - - - - 100 - - - - 8,560
Etano - - - - - - 100 - - - 15,235
Propano - - - - - - - 100 - - 21,809
Butano - - - - - - - - 100 - 28,365
Pentano - - - - - - - - - 100 34,912
Gas d’aria 27 5-10 3-14 55 1 0-3 - 0,9-1,33
Gas d’acqua 43 3 50 3 0,5 0,5 - 2,75
Gas d’acqua carburato 33 4 35 8 1 10 9 4,78
Gas di cokeria 7 2 53 3,5 0,5 2 32 5,22
Gas naturale secco - 0,5 - 3 - 95 1,5 8,63
Gas naturale umido - - - 1 - 68 31 11,10
GPL - - - - - - 2-0 90-10 10-90 0-1 22-27
Nei gas naturali sono spesso contenuti anche componenti acidi (H2S e CO2), azoto ed altri gas nobili come elio, e vapore d’acqua. Se il contenuto di composti acidi supera qualche unità percentuale si parla di “gas acidi” ed essi possono essere usati come risorsa naturale di derivati solforati. Se il contenuto di gas nobili supera qualche decimo di unità percentuale, la loro estrazione può diventare economicamente giustificata. Nella Tabella 3 è riportata la composizione di alcuni tipi di gas naturale estratti in diverse parti del mondo.
Tabella 3
Composizione di alcuni tipi di gas naturale
Componente G.N. “secco” (Ravenna, I)
G.N. “acido” (Lacq, F)
G.N. “umido” (Brega, Libia)
G.N. con Elio (Crossfield, USA)
CH4 C2H6 C3H8 C4H10 C5+ CO2 H2S N2 He
99,52 0,06 0,01
tracce
- 0,01
- 0,40
-
69,1 2,8 0,8 1,5 0,6 9,7
15,0 0,5 -
66,8 19,4 9,1 3,7 1,0 - - - -
80,60 6,45 2,30 1,23 1,27 6,13 1,06
- 0,96
I gas naturali devono essere opportunamente depurati e “stabilizzati” prima della loro immissione in gasdotti o metaniere. L’eliminazione delle particelle solide o delle gocce di liquido

17
trascinate dal gas si realizza semplicemente con mezzi meccanici (passaggio in labirinti, filtri, cicloni, ecc.). Il gas naturale deve essere anche essiccato accuratamente, per evitare condensazioni di acqua nei punti più freddi dei gasdotti. La disidratazione si esegue facendo passare i gas, ancora prima della decompressione, in torri di assorbimento alimentate con glicol di- o tri-etilenico o con miscele dei due. Questi fluidi assorbenti sono scelti in ragione della loro elevata affinità per l’acqua, del loro basso costo, della loro stabilità chimica, della bassa tendenza alla formazione di schiume e del basso potere solvente nei confronti del metano. Il contenuto di umidità può essere così ridotto fino a 40-120 mg/m3. Si possono usare anche assorbenti solidi, come SiO2, oppure, per i gas ad alta pressione, l’effetto Joule-Thompson. Per i gas naturali umidi, allorché si ricorra al raffreddamento fino a -90÷-100°C per l’eliminazione degli idrocarburi illuminanti, si rende necessaria una disidratazione più spinta che può realizzarsi con l’uso di letti di setacci molecolari che sono poi rigenerati per riscaldamento. La stabilizzazione dei gas naturali umidi (separazione degli idrocarburi condensabili) può eseguirsi per assorbimento in adatti oli di origine petrolifera, realizzato a bassa temperatura, oppure per raffreddamento con espansione fino a temperature di -90 ÷ -100°C. Il condensato viene poi frazionato per ottenere etano, propano, butani ed una frazione liquida detta casing-head gasoline.
I componenti acidi (H2S e CO2) possono essere rimossi con un gran numero di sistemi, la scelta dei quali dipende dal contenuto dei componenti acidi stessi che influisce sulla ottimizzazione economica del processo. Uno dei metodi più comuni è quello mediante assorbimento in “solvente chimico”, cioè in un solvente con caratteristiche debolmente basiche (ad es., una soluzione acquosa di monoetanolammina (MEA) o di dietanolammina (DEA)) capace di reagire chimicamente con la sostanza acida per dare un prodotto che, dopo separazione dal gas, può poi essere decomposto per semplice riscaldamento rigenerando il solvente che viene riciclato (V. Figura 1).
Figura 1
Desolforazione di gas naturale con soluzione di ammine. Il gas naturale viene trasportato e distribuito mediante gasdotti. A questo fine, il gas è compresso a circa 7 atm con compressori dislocati lungo il gasdotto a distanze calcolate in base alle perdite di carico. In vicinanza dei punti di distribuzione per usi civili, la pressione viene ridotta a pochi mm di Hg. In mancanza di gasdotti, il gas naturale viene trasportato in cisterne su autocarri o in metaniere, liquefatto a –160°C, ottenendo così una contrazione di volume di 600:1. La liquefazione del gas naturale è usata anche come sistema di stoccaggio in serbatoi in acciaio a doppia parete coibentata, generalmente fuori terra, per assorbire le punte di domanda. Il gas naturale ha soppiantato tutti gli altri gas combustibili descritti in precedenza, per la maggior parte degli impieghi, soprattutto per uso civile. Per la distribuzione del gas naturale in reti civiche, si rende necessaria una odorizzazione che viene ottenuta per aggiunta di piccole quantità (~20 mg/m3) di composti solforati come il tetraidrotiofene. Nell’industria, la produzione della necessaria energia termica viene effettuata anche bruciando tutti i gas combustibili ottenuti come

18
sottoprodotti di altre lavorazioni, per il loro minor costo. Il gas naturale è tuttavia considerato come una delle fonti di energia più pulite, per quanto riguarda l’impatto ambientale. Il gas naturale serve anche come importante materia prima per l’industria chimica. Da questo punto di vista, la maggior quantità di gas naturale è adoperata per produrre il gas di sintesi. Altre importanti utilizzazioni chimiche del gas naturale si hanno nella produzione di nerofumo, di acetilene, di acido cianidrico e di derivati clorurati. Processo di steam reforming del metano. Lo steam reforming del metano è oggi il processo più ampiamente usato per la produzione del gas di sintesi. Da un punto di vista formale, questo processo realizza una trasformazione esattamente inversa rispetto a quella del processo di produzione del gas naturale sintetico (SNG) accennato precedentemente. La giustificazione per la coesistenza, nell’ambito dell’industria chimica mondiale, di due processi formalmente opposti sta nella diversa disponibilità di risorse naturali. La produzione di SNG è economicamente conveniente solo in quei casi nei quali si abbia forte richiesta di metano e bassa disponibilità di gas naturale, nonché un’ampia disponibilità di carbone a basso prezzo. Questi casi sono ancora, mediamente, meno comuni a livello mondiale, e sono quelli nei quali il gas di sintesi viene ottenuto da sostanze carboniose piuttosto che da gas naturale.
Lo steam reforming del metano impiega catalizzatori al nickel che richiedono un contenuto di zolfo, nella alimentazione, inferiore a circa 2 ppm, per avere vita sufficientemente lunga. Oltre al metano, possono essere presenti anche idrocarburi superiori. È addirittura possibile, per ragioni di disponibilità, operare con frazioni liquide, come nafte. In ogni caso, la alimentazione deve essere accuratamente desolforata, prima dell’ingresso nel reformer. Ciò si realizza per passaggio su un letto catalitico di ossidi di Cobalto e Molibdeno (CoMox) con aggiunta di 2-5% di idrogeno, a 360-400°C, per convertire i composti solforati in H2S. Quest’ultimo è poi assorbito per passaggio attraverso due letti di ossido di zinco.
Dopo miscelamento con la quantità opportuna di vapore, la miscela viene quindi fatta reagire nel reformer su un catalizzatore di nickel con promotori alcalini, a 800-870°C e 25-35 atm. Le reazioni principali coinvolte nel processo sono:
CH4 + H2O ↔ CO + 3 H2 ∆H800°C = +54,3 Kcal/mole (1)(1)(1)(1)
CnHm + n H2O ↔ n CO + (n + ½ m) H2 ∆H800°C ≈ +50 Kcal/mole di C (2)(2)(2)(2)
Sia la (1), formalmente inversa della (6) del Cap. 3, che la (2) sono fortemente endotermiche ed il calore, a differenza di quanto avviene nei processi di produzione di gas di sintesi da carbone, viene fornito dall’esterno, attraverso le pareti del reattore. Secondo la stechiometria delle reazioni indicate sopra, la quantità di vapore d’acqua necessaria è di una mole per grammoatomo di carbonio. In realtà, il rapporto steam-to-carbon è normalmente compreso tra 3 e 5; i valori più alti si usano nei processi per la produzione di idrogeno nei quali, dopo il reforming, si realizza una conversione spinta dell’ossido di carbonio ad idrogeno secondo la reazione (4) del Cap. 3 (p. 14).
La miscela di gas desolforato e vapore è introdotta nel reattore di reforming primario, generalmente costituito da una batteria di tubi di acciaio speciale ad alto tenore di nickel, del diametro di circa 10 cm e di oltre 10 m di lunghezza, scaldati a fiamma diretta. Il catalizzatore contiene il 5-25% di nickel come NiO, supportato su alluminato di calcio e/o magnesio. Le velocità spaziali sono di 5000-8000 h-1. I gas caldi, all’uscita dalla sezione della fornace ove avviene la combustione, hanno una temperatura di 980-1050°C e passano nella zona di convezione della fornace stessa ove si realizza il preriscaldamento della alimentazione, la produzione di vapore (per l’impianto stesso e per altri impianti, o per la generazione di energia elettrica), il preriscaldamento dell’aria per la combustione, ecc. La loro temperatura, alla immissione in ciminiera deve essere ridotta a 150-170°C perché si realizzi un buon recupero termico.
I gas che lasciano il reformer contengono (su base secca) circa il 77% di H2, il 12% di CO, il 10% di CO2 e l’1% di CH4 (corrispondente ad una conversione superiore al 95%), oltre alle tracce

19
di azoto contenute nel gas naturale di partenza. L’eccesso di vapore d’acqua usato nella reazione serve a limitare la deposizione di coke sul catalizzatore, oltre che per aumentare la concentrazione di idrogeno e anidride carbonica a spese di quella di ossido di carbonio, secondo la (4) del Cap. 3.
Nei processi di produzione di idrogeno, i gas in uscita dal reformer sono inviati alla sezione di conversione ove si realizza la reazione (3) (identica alla (4) del Cap. 3, tranne che per la temperatura di riferimento):
CO + H2O ↔ CO2 + H2 ∆H370°C = -9,2 Kcal/mole (3)(3)(3)(3)
La reazione si realizza dapprima in uno stadio ad “alta” temperatura (circa 370°C) con un catalizzatore di ossido di ferro, con ossido di cromo come promotore, ed una velocità spaziale di 4000 h-1. I gas sono poi ulteriormente raffreddati fino a 200-215°C, con produzione di vapore, ed immessi nel convertitore a bassa temperatura ove un catalizzatore a base di ossidi di rame e zinco riduce il contenuto di CO a pochi decimi percentuali. L’assorbimento della anidride carbonica è effettuato dopo ulteriore raffreddamento dei gas a temperatura ambiente, usando adatti solventi, come una soluzione acquosa di carbonato di potassio, metiletanolammina (MEA), dimetiletanolammina (DMEA), ecc. Le tracce residue di ossido e biossido di carbonio possono essere eliminate, ove desiderato, per metanazione a 315°C, su un catalizzatore di nickel. La purificazione finale dell’idrogeno viene eseguita comunemente mediante assorbimento selettivo di tutte le altre molecole (più voluminose) come CO, CO2, N2, CH4, H2O, ecc., su setacci molecolari, attraverso cicli successivi di pressurizzazione e depressurizzazione, come mostrato in Figura 2. Con questo metodo si può produrre idrogeno con una purezza superiore al 99,9% e con una resa del 70-90%.
Figura 2
Processo di produzione di idrogeno puro mediante steam reforming, conversione, e purificazione per assorbimento su setacci molecolari (pressure swing absorption – PSA).
La nafta ed altri idrocarburi come propano e butano sono oggi usati come alimentazioni per lo steam reforming a gas di sintesi solo in caso di indisponibilità del gas naturale, dato che il loro prezzo è andato crescendo negli ultimi anni in quasi tutti i paesi occidentali. Il primo impianto di

20
steam reforming della nafta fu messo in esercizio dalla ICI, in Inghilterra, e molti altri impianti furono costruiti in seguito in tutto il mondo. Lavorando con nafta si ottiene una quantità maggiore di ossido di carbonio, a parità di idrogeno prodotto. Infatti, nella nafta si ha un numero di atomi di idrogeno per atomo di carbonio non lontano da due, mentre tale numero è pari a quattro per il metano. Qualora l’idrogeno puro sia il prodotto desiderato, si ha dunque una maggiore spesa per le operazioni di conversione e di separazione della CO2, oltreché per la materia prima, se si usa nafta. Processo di ossidazione parziale del gas naturale e di altre miscele di idrocarburi Il processo di ossidazione parziale di idrocarburi, e in particolare di metano, può risultare vantaggioso, rispetto allo steam reforming del gas naturale quando sia disponibile ossigeno a basso prezzo e, specialmente, quando occorrano gas di sintesi con elevato rapporto CO/H2. Il processo, del quale esistono diverse versioni messe a punto principalmente dalla Texaco e dalla Shell, si realizza iniettando l’idrocarburo preriscaldato, assieme ad ossigeno preriscaldato e vapore, in un apparecchio di combustione chiuso, nel quale, con una quantità di ossigeno substechiometrica, l’ossidazione parziale decorre a 1250-1500°C secondo la reazione:
CnHm + n/2 O2 → n CO + m/2 H2 (4)(4)(4)(4)
Il processo complessivo può essere suddiviso in tre fasi: 1) riscaldamento con cracking, 2) combustione, 3) conversione. Nella prima fase, l’idrocarburo subisce un repentino riscaldamento a causa del calore radiante emesso dalla fiamma della combustione e dalle pareti del reattore e crackizza in parte dando radicali liberi, metano e carbone. Nella fase di combustione, l’idrocarburo reagisce con l’ossigeno secondo la reazione:
CnHm + (n + m/4) O2 → n CO2 + m/2 H2O (5)(5)(5)(5)
Contemporaneamente, l’idrocarburo in eccesso reagisce con il vapore della alimentazione e con quello formato dalla (5) secondo la consueta reazione di steam reforming:
CnHm + n H2O ↔ n CO + (n + ½ m) H2 ∆H800°C ≈ +50 Kcal/mole di C (6)(6)(6)(6)
Nell’ultima fase, più lenta, una parte del carbone liberato dal cracking si consuma per reazione con la CO2 e con l’H2O. La composizione finale del gas prodotto dipende dal decorso della successiva reazione di conversione dell’ossido di carbonio ed acqua ad anidride carbonica e idrogeno, che dipende a sua volta dal modo con il quale si effettua il raffreddamento: se questo è molto veloce, il rapporto CO/H2 del gas si mantiene alto. Nei processi di ossidazione parziale, date le alte temperature, non è richiesto l’uso di catalizzatori. Inoltre, l’atmosfera riducente trasforma i composti solforati in idrogeno solforato e solfuro di carbonile, con un rapporto H2S/COS pari a circa 24. Per questo motivo, la desolforazione preliminare della alimentazione non è necessaria. La rimozione dei componenti acidi (H2S, COS, e CO2) dal gas di sintesi viene effettuata per lavaggio con solventi.
Processi di reforming combinato con ossidazione parziale
Per la sintesi del metanolo, è richiesta una composizione del gas di sintesi con CO/H2 ≈ 1:2, mentre, come abbiamo visto, il processo di reforming del metano (che fornisce almeno l’80% del gas di sintesi usato a livello mondiale per la produzione di metanolo) produce miscele con CO/H2 ≈ 1:3. In alcuni impianti di produzione di metanolo, l’eccesso di idrogeno viene allontanato con lo spurgo e bruciato per produrre energia o inviato all’impianto di produzione di ammoniaca. In altri impianti si aggiunge anidride carbonica al gas di alimentazione, per portarne la composizione ai valori richiesti dalla stechiometria. Dato che, come mostrato nel paragrafo precedente, la produzione di gas di sintesi per ossidazione parziale fornisce miscele con rapporto CO/H2 più alto,

21
sono stati commercializzati anche processi di “reforming combinato” nei quali il reforming primario con vapore, con apporto energetico a fiamma diretta, è seguito da un reforming secondario realizzato per ossidazione parziale (V. Figura 3). Il reattore secondario è un recipiente adiabatico rivestito internamente in materiale refrattario, riempito con un catalizzatore di Nickel resistente alle alte temperature, nel quale si inietta una appropriata quantità di ossigeno puro in modo da avere un rapporto complessivo CO/H2 ≈ 1:2.
Figura 3
Schema di reforming combinato. Usando questa soluzione, oltre a risparmiare combustibile per il reforming primario ed ottenere una composizione ottimale del gas di sintesi, è possibile far lavorare il reformer primario a pressioni più alte e a temperature più basse, perché il metano residuo può essere ulteriormente trasformato alle alte temperature raggiunte nel reformer secondario, realizzando così una economia complessiva nei consumi di energia per la compressione. Una estensione del concetto di reforming combinato è rappresentata dal processo con reformer riscaldato con i gas del reforming secondario. Il reformer primario è in realtà uno scambiatore di calore (GHR – gas
heated reformer), con il catalizzatore contenuto in doppi tubi a baionetta (V. Figura 4). Il gas naturale desolforato ed il vapore entrano dalla testa del reattore e scendono verso il basso nella sezione anulare dei doppi tubi (ove è contenuto il catalizzatore) per risalire poi verso l’alto nei tubi interni ed uscire lateralmente dalla testa dell’apparecchio (effluente primario). L’effluente primario è alimentato al reformer secondario dal quale i gas escono a temperatura elevata e sono alimentati al reattore primario dal basso, per lambire le pareti esterne dei doppi tubi scaldandoli.
Usi del gas di sintesi
L’impiego più importante del gas di sintesi consiste nella produzione di ammoniaca. Questa industria consuma circa il 5% di tutto il gas naturale e del gas associato prodotto nel mondo. Malgrado che l’ammoniaca sia un composto inorganico, essa si produce a partire da risorse naturali
Ossigeno Combustibile
Gas naturale Desolforazione Reforming primario
Reforming secondario
Gas di sintesi
Figura 4
Gas heated reformer della ICI.

22
organiche e si impiega per la sintesi di numerosi prodotti organici. Il secondo prodotto, in ordine di importanza, ottenuto dal gas di sintesi, è il metanolo. La dimensione di questa industria, in termini di gas naturale consumato, è di circa un quinto, rispetto a quella dell’ammoniaca. Occorre notare, però, che il 20% circa del gas di sintesi usato per produrre metanolo è ottenuto a partire da carbone. Il metanolo è impiegato in misura sempre crescente per produrre MTBE, oltreché aldeide formica, acido acetico e derivati. Il metanolo è anche usato, in Sud Africa, come componente delle benzine.
_______________________

23
Capitolo 3
IL PETROLIO ED I PRODOTTI PETROLIFERI Il Petrolio
Il petrolio è una sostanza fossile liquida, più o meno densa e viscosa, di colore variabile dal bruno al nero, costituita da una miscela estremamente complessa di composti, prevalentemente idrocarburici. Gli americani usano spesso il termine petroleum per indicare tutti i fossili idrocarburici, sia quelli liquidi (crude oil) che quelli gassosi (natural gas), che quelli solidi (bitumen). Il petrolio (crude oil) è spesso identificato anche come petrolio grezzo o semplicemente “grezzo”.
Circa le origini del petrolio, sono state avanzate diverse teorie. Secondo l’ipotesi che gode oggi di maggior credito, il petrolio è il prodotto delle trasformazioni chimiche, fisiche e microbiologiche subite dai depositi di sostanze organiche costituite dai resti di organismi sia animali che vegetali. Che alla formazione dei giacimenti petroliferi abbiano contribuito in maniera consistente gli organismi animali, includenti lo zooplancton e lo zoobenton, oltre a quelli vegetali, includenti il fitoplancton ed il fitobenton generati per fotosintesi in ambienti lacustri o in mare, è dimostrato sia dalla presenza nel grezzo di sostanze otticamente attive tipiche dei tessuti animali, sia di elementi metallici come il nichel, il vanadio ed il ferro, anch’essi caratteristici dei sistemi proteici animali, sia, infine, dalla prevalenza degli idrocarburi paraffinici con numero dispari di atomi di carbonio, spiegabile attraverso processi di decarbossilazione di acidi grassi animali, i quali contengono sempre un numero pari di atomi di carbonio.
Durante il processo di fossilizzazione, i sedimenti organici, attraverso reazioni di decarbossilazione indotte da agenti anaerobici, subiscono una progressiva riduzione del contenuto di ossigeno, trasformandosi, nelle “rocce madri”, in una sostanza organica solida detta kerogen, dalla quale si formano, per successive trasformazioni, prodotti gassosi e liquidi, lasciando un residuo carbonioso grafitico. I liquidi ed i gas si spostano quindi, per effetto della pressione, attraverso strati permeabili (arenarie), fino ad incontrare “trappole”, costituite da strati impermeabili, in genere argillosi, ove si raccolgono dando luogo ai giacimenti di petrolio e/o di gas naturale (Fig. 1).
Figura 1 Schema di trappola petrolifera di tipo anticlinale.
L’estrazione del grezzo dai giacimenti si esegue mediante trivellazione con un’asta rotante, costituita da una serie allungabile di tubi in acciaio portante all’estremità inferiore uno “scalpello” a diamanti o a rulli conici, e fissata in alto ad una “tavola rotante” alloggiata su una incastellatura metallica (derrik). Attraverso i tubi che compongono l’asta rotante viene pompata acqua che, fuoruscita dallo scalpello, risale nell’intercapedine tra i tubi e le pareti del pozzo portando in superficie il materiale lapideo abraso dallo scalpello sotto forma di fango fluido. In genere, quando lo scalpello raggiunge il giacimento, la pressione del gas presente nel pozzo è sufficiente a far
Calcare
Argilla
Argilla
Gas in arenaria
Acqua in arenaria
Petrolio in arenaria

24
sgorgare il grezzo in superficie. Comunque, verso la fine dello sfruttamento, è sempre necessario aspirare il grezzo mediante pompe a stantuffo azionate dai tipici bilancieri che caratterizzano i campi di estrazione. Può anche essere necessario fluidificare il greggio per immissione di gasolio, di acqua o, addirittura di vapore, allo scopo di spingere il più possibile lo sfruttamento. Con sistemi di estrazione moderni, basati sull’uso di queste tecniche, è stato possibile rimettere in funzione pozzi precedentemente abbandonati. La disponibilità di grezzo nei giacimenti varia in intervalli molto ampi: i giacimenti con oltre 80.000.000 m3 (circa 500 milioni di barili) di grezzo recuperabile sono chiamati “giganti”. Il grezzo estratto deve essere sottoposto in loco ad una serie di trattamenti per la separazione dell’acqua che lo accompagna, in parte sotto forma di emulsione, e dei solidi sospesi. Esso deve essere inoltre “stabilizzato”, cioè separato dal così detto “gas associato”, costituito dagli idrocarburi leggeri in esso disciolti, che potrebbero dar luogo ad inconvenienti notevoli liberandosi durante il trasporto in oleodotti o petroliere. Tutte queste operazioni si eseguono mediante decantazione, centrifugazione, percolazione attraverso letti sabbiosi, riscaldamento, ecc. Il trasporto del grezzo agli impianti di raffinazione si effettua in oleodotti (tubi in acciaio del diametro di 50-90 cm e lunghezza talvolta superiore ai 3000 Km, nei quali il grezzo viene fatto fluire da stazioni di pompaggio dislocate lungo il percorso con velocità di 1-2 m/s) o in petroliere. Queste sono capaci di trasportare fini a 350.000 t, distribuite in adatti scomparti isolati l’uno dall’altro. Le unità di misura più in uso per il grezzo sono la tonnellata, corrispondente ad un volume un po’ superiore al m3, ed il barile:
1 bbl = 159 L
Composizione del grezzo
Il grezzo è costituito da una miscela complessa di composti tra i quali prevalgono gli idrocarburi. Esso contiene anche composti organici solforati, azotati e ossigenati, piccole quantità di composti organometallici, oltre a quantità variabili di gas disciolti e di acqua contenente sali. La composizione elementare dei grezzi varia normalmente entro i seguenti limiti:
C = 83-87% H = 11,4-14% S = 0,06-8% N = 0,01-1,8% O = 0,05-3%.
Si noti che il rapporto ponderale tra H e C corrisponde ad un rapporto tra grammoatomi variabile tra 1,59 e 2,02. La composizione media degli idrocarburi dei grezzi, dunque, non si discosta troppo da quella corrispondente al gruppo metilenico CH2. Lo zolfo, può essere presente nel grezzo sotto forma di H2S, di composti organici, o di zolfo libero disciolto (se il contenuto totale supera il 5% circa). Molti composti organici solforati sono termicamente instabili e possono decomporsi liberando H2S all’atto della distillazione. Si noti che ad un contenuto di zolfo del 2% corrisponde un contenuto in composti solforati di circa il 20%. Lo zolfo è di solito contenuto in quantità maggiori nei componenti più pesanti del petrolio e lo si ritrova quindi in quantità maggiori nelle frazioni più altobollenti e, ancora di più, nel residuo. La presenza di zolfo nei prodotti petroliferi è dannosa principalmente perché porta alla formazione di SO2 nei processi di combustione. I composti solforati, inoltre, avvelenano i catalizzatori metallici impiegati in alcuni processi di raffinazione. Lo zolfo può essere eliminato dalle correnti di raffineria mediante idrogenazione ad H2S (cf. Cap. 6, p. 5). Questa operazione, tuttavia, non è sempre economicamente sostenibile. I composti organici solforati più comunemente presenti nel grezzo sono i mercaptani, i solfuri ed i tiofeni. I primi sono i composti organici contenenti lo zolfo sotto forma di gruppo funzionale -SH: essi assomigliano strutturalmente agli alcoli, dai quali formalmente si ottengono per sostituzione di un atomo di ossigeno con uno di zolfo. Nei solfuri e nei tiofeni lo zolfo è invece legato a due atomi di carbonio della stessa molecola. I tiofeni sono molecole più o meno complesse contenenti l’anello a 5 atomi (4 di C ed 1 di S) con doppia insaturazione tipico del tiofene (CH)4S.
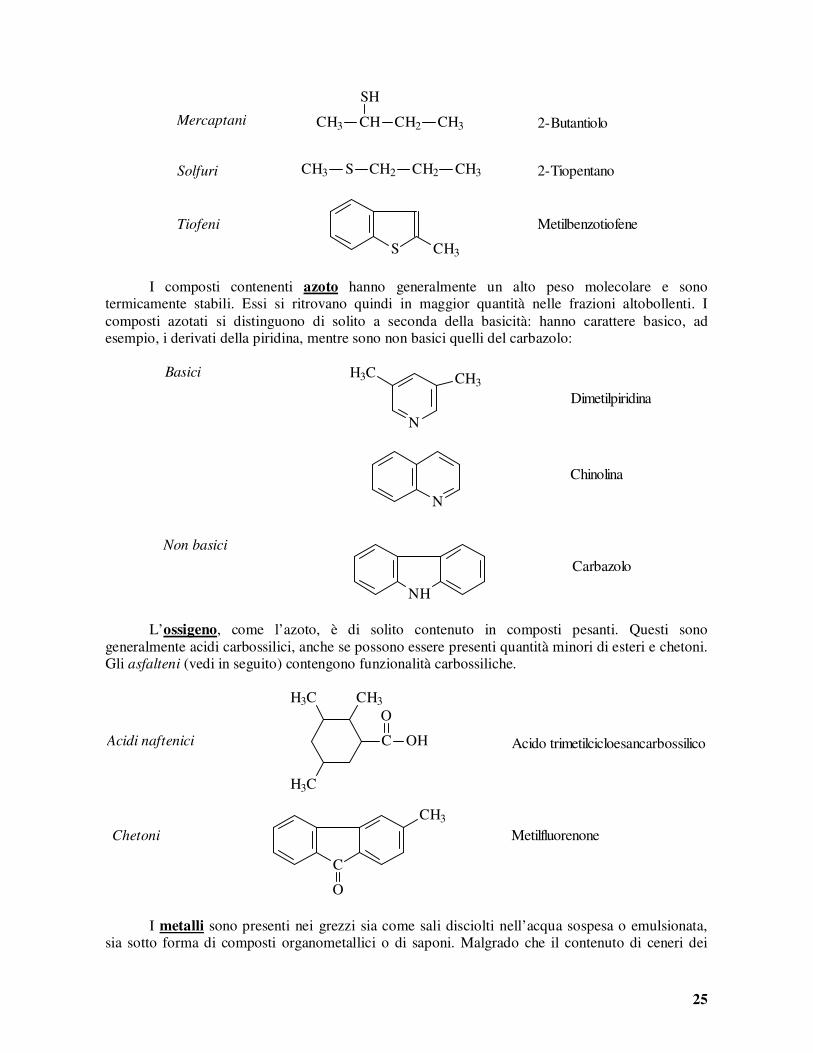
25
I composti contenenti azoto hanno generalmente un alto peso molecolare e sono termicamente stabili. Essi si ritrovano quindi in maggior quantità nelle frazioni altobollenti. I composti azotati si distinguono di solito a seconda della basicità: hanno carattere basico, ad esempio, i derivati della piridina, mentre sono non basici quelli del carbazolo:
L’ossigeno, come l’azoto, è di solito contenuto in composti pesanti. Questi sono generalmente acidi carbossilici, anche se possono essere presenti quantità minori di esteri e chetoni. Gli asfalteni (vedi in seguito) contengono funzionalità carbossiliche.
I metalli sono presenti nei grezzi sia come sali disciolti nell’acqua sospesa o emulsionata, sia sotto forma di composti organometallici o di saponi. Malgrado che il contenuto di ceneri dei
Mercaptani
Solfuri
Tiofeni
CH3 CH CH2
SH
CH3 2-Butantiolo
CH3 S CH2 CH2 CH3 2-Tiopentano
S CH3
Metilbenzotiofene
Non basici
Chinolina
Carbazolo
Basici
Dimetilpiridina
N
H3C CH3
N
NH
Chetoni Metilfluorenone
Acidi naftenici Acido trimetilcicloesancarbossilico
H3C CH3
H3C
C
O
OH
C
CH3
O

26
grezzi sia sempre molto piccolo (0,001-0,03%), è stata accertata la presenza di tracce di composti organometallici del V, del Ni, del Fe, come ad es. le vanadilporfirine. Le famiglie di idrocarburi presenti nei grezzi sono: 1) n-paraffine e isoparaffine, presenti nei grezzi in maggior quantità (45-80%); 2) cicloparaffine (dette anche nafteni), che sono di solito i secondi maggiori componenti (20-50%); 3) aromatici, che non superano, salvo eccezioni, il 10%.
Sia le cicloparaffine che gli aromatici contengono spesso uno o più anelli, oltre a gruppi alchilici. Gli idrocarburi insaturi come le olefine e le diolefine sono praticamente assenti nei grezzi.
La complessità della composizione dei grezzi è tale che solo i componenti più bassobollenti possono essere separati in forma pura. La Tabella 1 riporta la composizione di un tipico grezzo, limitatamente agli idrocarburi con punto di ebollizione minore o uguale a quello del toluene.
Tabella 1
Idrocarburo
Teb (°C) Contenuto (% vol)
Metano Etano Propano Isobutano n-Butano 2-Metilbutano n-Pentano Ciclopentano 2,2-Dimetilbutano 2,3-Dimetilbutano 2-Metilpentano 3-Metilpentano n-Esano Metilciclopentano 2,2-Dimetilpentano Benzene 2,4-Dimetilpentano Cicloesano 1,1-Dimetilciclopentano 2,3-Dimetilpentano 2-Metilesano 1,cis-3-Dimetilciclopentano 1,trans-3-Dimetilciclopentano 3-Metilesano 1,trans-2-Dimetilciclopentano 3-Etilpentano n-Eptano Metilcicloesano Etilciclopentano 1,1,3-Trimetilciclopentano 2,2-Dimetilesano 2,5-Dimetilesano 1,trans-2,cis-4-Trimetilciclopentano 2,4-Dimetilesano 2,2,3-Trimetilpentano 1,trans-2,cis-3-Trimetilciclopentano Toluene
-161,5 -88,6 -42,1 -11,7 -0,5 27,8 36,1 49,3 49,7 58,0 60,3 63,3 68,7 71,8 79,2 80,1 80,5 80,7 87,8 89,8 90,0 90,8 91,7 91,8 91,9 93,5 98,4
100,9 103,5 104,9 106,8 109,1 109,3 109,4 109,8 110,2 110,6
- 0,06 0,11 0,14 0,79 0,77 1,43 0,05 0,04 0,08 0,37 0,35 1,80 0,87 0,02 0,15 0,08 0,71 0,16 0,15 0,73 0,87 0,21 0,51 0,48 0,06 2,31 1,60 0,16 0,30 0,01 0,06 0,22 0,06 0,004 0,26 0,51

27
I dati nella Tabella dimostrano chiaramente che i punti di ebollizione degli idrocarburi si avvicinano sempre di più l’uno all’altro al crescere del numero di atomi di carbonio perché, contemporaneamente, cresce in modo esponenziale il numero dei possibili isomeri. La Tabella dimostra anche che il contenuto di ciascun idrocarburo nel grezzo è estremamente piccolo. Di conseguenza, per gli idrocarburi più pesanti, la separazione in forma pura diventa addirittura impossibile, oltreché antieconomica,. La Tabella mostra infine che gli idrocarburi n-paraffinici sono contenuti in percentuale maggiore di tutti gli altri.
La classificazione dei diversi tipi di grezzo viene fatta normalmente secondo due principali criteri: il primo, particolarmente importante sul piano commerciale in quanto parametro di riferimento del prezzo ufficiale dei grezzi, si riferisce alla loro densità ed al loro contenuto in zolfo; il secondo riguarda il contenuto relativo delle tre famiglie di idrocarburi, che può essere ricavato ancora da misure di densità. La densità dei grezzi e delle loro frazioni viene espressa convenzionalmente in gradi API (American Petroleum Institute):
1°API = (141,5/ps) – 131,5
dove ps indica il peso specifico misurato a 60°F (15,6°C).
Una sostanza con peso specifico pari ad 1 ha una densità API di 10°. I pesi specifici dei più comuni grezzi sono normalmente compresi tra 0,80 e 0,95, corrispondenti a 45 e 17°API, come mostrato nella Tabella 2 che riporta la densità ed il contenuto in zolfo di alcuni grezzi tipici.
Tabella 2
Densità e contenuto in zolfo di alcuni grezzi.
Paese
Grezzo ps °API %S
Arabia Saudita
Kuwait Iraq Iran
Algeria Libia UK
Venezuela
Messico Russia USA
Arabian light Arabian heavy
Kuwait Kirkuk
Iranian heavy Saharian blend
Brega Brent blend Bachaquero
T. Juana light Maya
Export grade W.T.I.
0,8550 0,8927 0,8708 0,8448 0,8708 0,8063 0,8156 0,8348 0,9529 0,8708 0,9218 0,8654 0,8063
34 27 31 36 31 44 42 38 17 31 22 32 44
1,7 2,8 2,5 2,0 1,6 0,1 0,2 0,3 2,4 1,2 3,4 0,1 0,7
La Tabella indica che, in base alla densità, i grezzi sono classificati in leggeri e pesanti; in
generale, i grezzi leggeri sono anche quelli con contenuto di zolfo più basso e con pregio commerciale maggiore. La “leggerezza” di un grezzo dipende sia dal loro maggiore contenuto in frazioni leggere, sia, come spiegato nel seguito, dal loro carattere prevalentemente paraffinico.
Negli ultimi anni, hanno acquistato crescente interesse per le raffinerie anche grezzi molto pesanti o, addirittura, bitumi (derivanti da giacimenti di sabbie o scisti bituminosi). Si classificano come “pesanti” i grezzi con densità API compresa tra 30 e 10°, come “extra pesanti” quelli con densità API minore di 10° e viscosità inferiore a 10.000 cp, come “bitumi” quelli con densità API minore di 10° e viscosità superiore a 10.000 cp.
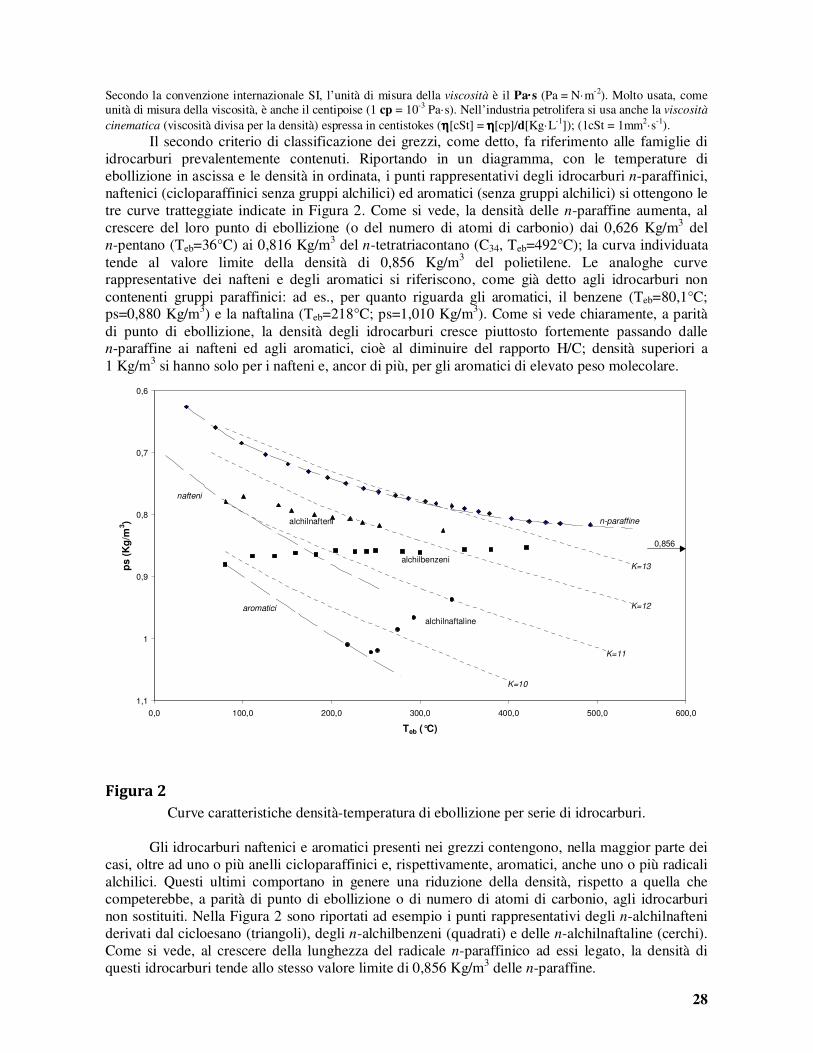
28
Secondo la convenzione internazionale SI, l’unità di misura della viscosità è il Pa·s (Pa = N·m-2). Molto usata, come unità di misura della viscosità, è anche il centipoise (1 cp = 10-3 Pa·s). Nell’industria petrolifera si usa anche la viscosità
cinematica (viscosità divisa per la densità) espressa in centistokes (ηηηη[cSt] = ηηηη[cp]/d[Kg·L-1]); (1cSt = 1mm2·s-1). Il secondo criterio di classificazione dei grezzi, come detto, fa riferimento alle famiglie di
idrocarburi prevalentemente contenuti. Riportando in un diagramma, con le temperature di ebollizione in ascissa e le densità in ordinata, i punti rappresentativi degli idrocarburi n-paraffinici, naftenici (cicloparaffinici senza gruppi alchilici) ed aromatici (senza gruppi alchilici) si ottengono le tre curve tratteggiate indicate in Figura 2. Come si vede, la densità delle n-paraffine aumenta, al crescere del loro punto di ebollizione (o del numero di atomi di carbonio) dai 0,626 Kg/m3 del n-pentano (Teb=36°C) ai 0,816 Kg/m3 del n-tetratriacontano (C34, Teb=492°C); la curva individuata tende al valore limite della densità di 0,856 Kg/m3 del polietilene. Le analoghe curve rappresentative dei nafteni e degli aromatici si riferiscono, come già detto agli idrocarburi non contenenti gruppi paraffinici: ad es., per quanto riguarda gli aromatici, il benzene (Teb=80,1°C; ps=0,880 Kg/m3) e la naftalina (Teb=218°C; ps=1,010 Kg/m3). Come si vede chiaramente, a parità di punto di ebollizione, la densità degli idrocarburi cresce piuttosto fortemente passando dalle n-paraffine ai nafteni ed agli aromatici, cioè al diminuire del rapporto H/C; densità superiori a 1 Kg/m3 si hanno solo per i nafteni e, ancor di più, per gli aromatici di elevato peso molecolare.
Figura 2
Curve caratteristiche densità-temperatura di ebollizione per serie di idrocarburi. Gli idrocarburi naftenici e aromatici presenti nei grezzi contengono, nella maggior parte dei
casi, oltre ad uno o più anelli cicloparaffinici e, rispettivamente, aromatici, anche uno o più radicali alchilici. Questi ultimi comportano in genere una riduzione della densità, rispetto a quella che competerebbe, a parità di punto di ebollizione o di numero di atomi di carbonio, agli idrocarburi non sostituiti. Nella Figura 2 sono riportati ad esempio i punti rappresentativi degli n-alchilnafteni derivati dal cicloesano (triangoli), degli n-alchilbenzeni (quadrati) e delle n-alchilnaftaline (cerchi). Come si vede, al crescere della lunghezza del radicale n-paraffinico ad essi legato, la densità di questi idrocarburi tende allo stesso valore limite di 0,856 Kg/m3 delle n-paraffine.
0,6
0,7
0,8
0,9
1
1,1
0,0 100,0 200,0 300,0 400,0 500,0 600,0
Teb (°C)
ps (
Kg
/m3)
0,856
n-paraffine
nafteni
aromatici
alchilnafteni
alchilbenzeni
alchilnaftaline
K=10
K=11
K=12
K=13

29
Il fattore di caratterizzazione K (detto anche Kuop), definito empiricamente dalla relazione
dove ps è la densità misurata a 60°F (15,6°C) e Teb è espressa in gradi Rankin (°R=°F+459,4), individua delle curve, pure riportate in Figura 2, il cui andamento è simile a quello delle curve tipiche delle diverse famiglie “pure” di idrocarburi (n-paraffine, nafteni e aromatici). La natura chimica di un idrocarburo o di una miscela di idrocarburi può essere definita in modo approssimato dal suo valore di K. Una frazione di grezzo cui competa un fattore di caratterizzazione K compreso tra 9,5 e 10,5 è considerata aromatica; tra 10,5 e 11,5 naftenica; tra 11,5 e 12 intermedia; tra 12 e 13 paraffinica. Una classificazione dei grezzi in base al valore del fattore di caratterizzazione K, può essere significativa sul piano pratico perché ne individua il valore commerciale, ma è piuttosto grossolana, non solo per la difficoltà di definire una temperatura di ebollizione media, ma anche perché il fattore di caratterizzazione delle frazioni bassobollenti è spesso diverso (in genere maggiore) rispetto a quello delle frazioni altobollenti dello stesso grezzo. Un metodo di classificazione più affidabile e più utile si basa sulla caratterizzazione delle frazioni di ciascun grezzo. Molto spesso, la caratterizzazione si fa con riferimento alle così dette frazioni chiave. Secondo il Bureau of Mines, si considerano ad esempio due frazioni chiave: la n° 1, con intervallo dei punti di ebollizione a pressione atmosferica compreso tra 250 e 275°C; la n° 2, con punti di ebollizione tra 275 e 300°C alla pressione di 40 mm Hg. Attraverso misure di densità eseguite su queste due frazioni chiave, i grezzi possono quindi essere classificati come mostrato nella Tabella 3.
Tabella 3
Grezzo
Frazione n° 1 Frazione n° 2
ps °API ps °API
Paraffinico Intermedio Naftenico
<0,825 0,825-0,860
>0,860
>40 33-40 <33
<0,876 0,876-0,934
>0,934
>30 20-30 <20
La definizione della temperatura di ebollizione media di una frazione, da utilizzare per il calcolo del fattore di caratterizzazione K, si fa a partire dalla curva di distillazione determinata secondo un metodo standard per tale frazione. Il metodo più semplice e rapido è quello definito dalla norma ASTM D 86. Il metodo è rappresentato schematicamente in Figura 3, assieme alla curva di distillazione ottenuta.
ps
R)(TK
3 eb °=
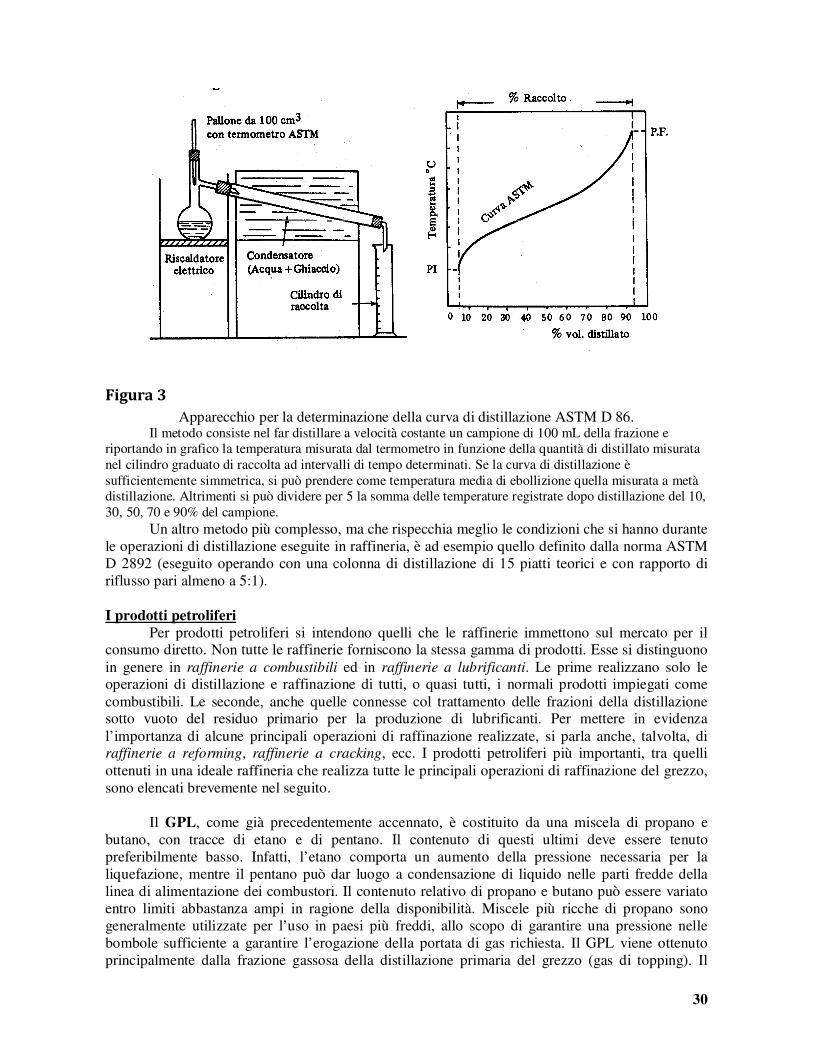
30
Figura 3
Apparecchio per la determinazione della curva di distillazione ASTM D 86. Il metodo consiste nel far distillare a velocità costante un campione di 100 mL della frazione e riportando in grafico la temperatura misurata dal termometro in funzione della quantità di distillato misurata nel cilindro graduato di raccolta ad intervalli di tempo determinati. Se la curva di distillazione è sufficientemente simmetrica, si può prendere come temperatura media di ebollizione quella misurata a metà distillazione. Altrimenti si può dividere per 5 la somma delle temperature registrate dopo distillazione del 10, 30, 50, 70 e 90% del campione. Un altro metodo più complesso, ma che rispecchia meglio le condizioni che si hanno durante le operazioni di distillazione eseguite in raffineria, è ad esempio quello definito dalla norma ASTM D 2892 (eseguito operando con una colonna di distillazione di 15 piatti teorici e con rapporto di riflusso pari almeno a 5:1). I prodotti petroliferi
Per prodotti petroliferi si intendono quelli che le raffinerie immettono sul mercato per il consumo diretto. Non tutte le raffinerie forniscono la stessa gamma di prodotti. Esse si distinguono in genere in raffinerie a combustibili ed in raffinerie a lubrificanti. Le prime realizzano solo le operazioni di distillazione e raffinazione di tutti, o quasi tutti, i normali prodotti impiegati come combustibili. Le seconde, anche quelle connesse col trattamento delle frazioni della distillazione sotto vuoto del residuo primario per la produzione di lubrificanti. Per mettere in evidenza l’importanza di alcune principali operazioni di raffinazione realizzate, si parla anche, talvolta, di raffinerie a reforming, raffinerie a cracking, ecc. I prodotti petroliferi più importanti, tra quelli ottenuti in una ideale raffineria che realizza tutte le principali operazioni di raffinazione del grezzo, sono elencati brevemente nel seguito.
Il GPL, come già precedentemente accennato, è costituito da una miscela di propano e butano, con tracce di etano e di pentano. Il contenuto di questi ultimi deve essere tenuto preferibilmente basso. Infatti, l’etano comporta un aumento della pressione necessaria per la liquefazione, mentre il pentano può dar luogo a condensazione di liquido nelle parti fredde della linea di alimentazione dei combustori. Il contenuto relativo di propano e butano può essere variato entro limiti abbastanza ampi in ragione della disponibilità. Miscele più ricche di propano sono generalmente utilizzate per l’uso in paesi più freddi, allo scopo di garantire una pressione nelle bombole sufficiente a garantire l’erogazione della portata di gas richiesta. Il GPL viene ottenuto principalmente dalla frazione gassosa della distillazione primaria del grezzo (gas di topping). Il

31
butano contenuto nel gas di topping ha anche utilizzazioni alternative importanti come materia prima per operazioni di isomerizzazione, nonché come “blending stock” per migliorare la volatilità delle benzine, specie nei paesi più freddi. Il propano ha anch’esso impieghi alternativi, ad esempio come solvente per l’eliminazione delle resine e degli asfalteni dagli oli lubrificanti.
Le benzine sono le miscele di idrocarburi, con numero di atomi di carbonio generalmente
compreso tra 5 e 12 (Teb compreso tra 30 e 220°C), che si utilizzano come carburanti per motori a scoppio. Le caratteristiche più importanti delle benzine sono la volatilità, determinabile dalle curve di distillazione, il numero d’ottano, la suscettività, la sensitività e la stabilità chimica. La volatilità deve essere opportunamente regolata all’atto del “blending” (miscelazione dei diversi blending stocks per ottenere la formulazione desiderata) in modo da garantire una buona vaporizzazione anche a motore freddo.
Il numero d’ottano è una misura del carattere indetonante di una benzina. Nei motori a scoppio, come è noto, la combustione della miscela deve avvenire in modo graduale, anche se molto veloce, per propagazione di un fronte di fiamma generato dalla scintilla che viene fatta scoccare in un istante accuratamente programmato, in vicinanza del tempo corrispondente al “punto morto superiore” del moto del pistone (massima compressione della miscela). L’indesiderata “detonazione” si verifica quando una parte della miscela benzina-aria si incendia spontaneamente per compressione, senza attendere il fronte di fiamma generato dalla scintilla. Essa è generalmente udibile al di sopra del normale rumore del motore e causa un impulso di pressione che agisce sul pistone prima che questo abbia raggiunto il punto morto superiore, con conseguente perdita di potenza. La tendenza di una benzina a detonare o, viceversa, il suo carattere indetonante, dipendono dalla struttura chimica degli idrocarburi che la compongono, oltreché dalle condizioni di funzionamento del motore.
Per quanto riguarda la struttura chimica degli idrocarburi, può dirsi in generale che idrocarburi con geometria globulare della molecola presentano un elevato potere indetonante, mentre, al contrario, idrocarburi con geometria molecolare fortemente anisometrica (cioè con dimensioni molecolari spiccatamente dissimili nelle tre dimensioni dello spazio) hanno elevata tendenza alla detonazione. Ad esempio, l’isottano (2,2,4-trimetilpentano), ha elevato potere indetonante, mentre il n-eptano non ha questo carattere. Questi due idrocarburi sono stati scelti per definire una scala empirica per la misura del carattere indetonante delle benzine. Si dice che una benzina ha numero di ottano 98 quando il suo comportamento nei confronti del fenomeno della detonazione è uguale a quello di una miscela contenente il 98% di isottano ed il 2% di n-eptano.
CH3 C
CH3
CH3
CH2 CH
CH3
CH3 CH3 CH2 CH2 CH3 CH2 CH2 CH3
2,2,4-Trimetilpentano n-Eptano

32
L’anisometria molecolare è in genere elevata per le n-paraffine; pertanto, le benzine ottenute per semplice frazionamento di grezzi paraffinici hanno basso numero di ottano. Il numero di ottano degli idrocarburi n-paraffinici cresce fortemente al diminuire del loro numero di atomi di carbonio (V. Tabella 4). Per questa ragione, il metano ed il GPL possono essere usati per l’alimentazione di motori a scoppio senza che sia richiesta l’aggiunta di agenti antidetonanti.
Tabella 4
Numero di ottano di alcuni idrocarburi.
Idrocarburo
N.O.
Metano Propano n-Butano n-Pentano n-Esano n-Eptano Benzene Toluene Etilbenzene Xileni
125 111 103 62 25 0
113 120 107 140
Gli idrocarburi cicloparaffinici e più ancora le olefine a catena lineare hanno numeri di ottano più alti di quelli delle n-paraffine con ugual numero di atomi di carbonio. Tuttavia, la presenza di olefine nelle benzine ne può diminuire la stabilità chimica (possibile formazione di polimeri) e richiede l’aggiunta di adatti agenti stabilizzanti (alchilfenoli e ammine aromatiche come antiossidanti e inibitori di polimerizzazione). Gli idrocarburi iso-paraffinici hanno N.O. tanto più alto quanto maggiore è il loro grado di ramificazione. Anche gli idrocarburi aromatici hanno N.O. elevato (V. Tabella 4). Per quanto riguarda le condizioni di funzionamento del motore, è evidente che, a parità di struttura chimica degli idrocarburi, il pericolo di detonazione aumenta al crescere del rapporto di compressione del motore. A questo proposito è utile rilevare che negli ultimi decenni l’industria automobilistica, con lo scopo di perseguire una politica di riduzione dei consumi di carburante e, ancor di più, di limitare l’incidenza delle imposizioni fiscali, che in Italia sono proporzionate alla cilindrata delle autovetture, si è mossa verso una graduale riduzione delle cilindrate dei motori, aumentandone parallelamente i rapporti di compressione per non diminuirne la potenza. Ciò ha portato ad una sempre crescente esigenza di disporre di benzine con numero di ottano accresciuto. La tendenza alla detonazione di una data benzina aumenta in generale anche all’aumentare della severità di esercizio del motore: una benzina che non detona in regime di funzionamento blando, può dar luogo a detonazione quando il motore è portato a numero di giri elevato. La determinazione del numero di ottano di una benzina si fa mediante l’uso di adatti motori secondo due principali metodi standardizzati: il metodo Research simula le condizioni di funzionamento su strada a bassa severità; il metodo Motor simula condizioni di funzionamento più severe. Il numero di ottano determinato con i due metodi è indicato, rispettivamente, come RON (Research Octane Number) e come MON (Motor Octane Number). Da quanto detto precedentemente, risulta chiaro che, per una data benzina, il RON risulta sempre più elevato del MON. La differenza RON – MON rappresenta un indice della variazione del potere indetonante di una benzina al variare delle condizioni di esercizio del motore. Tale differenza è definita come sensitività della benzina. Quanto minore è la sensitività, tanto migliore è la benzina. Negli Stati Uniti, è molto usato, come parametro per definire il potere indetonante delle benzine il così detto “numero di ottano strada”, dato dalla media (RON + MON)/2. In Italia, invece, il riferimento viene fatto generalmente al RON.

33
Le benzine immesse al consumo per i diversi impieghi (benzine per autotrazione, benzine avio, benzine per agricoltura, ecc.) sono prodotte mediante miscelazione (blending) dei diversi blending stocks derivanti dalle diverse operazioni effettuate in raffineria (benzine di 1a distillazione, di cracking, di idrocracking, di reforming, di alchilazione, di isomerizzazione, di polimerizzazione, ecc.) e sono “formulate” mediante aggiunta di opportuni agenti (antidetonanti, stabilizzanti, ecc.). Particolare importanza rivestono gli agenti antidetonanti che servono ad aumentare il N.O. “clear” di una benzina (cioè quello proprio della benzina senza additivi) portandolo al valore fissato dalle specifiche per i diversi tipi di benzina commerciali. L’agente antidetonante al quale si è fatto più largamente ricorso, fino ad oggi, è il piombo tetraetile Pb(Et)4 (indicato nei testi inglesi come TEL). L’aggiunta di quantità relativamente piccole (0,3-0,8 mL/L)di TEL alle benzine aumenta piuttosto fortemente il loro carattere indetonante. Non tutte le benzine, tuttavia, subiscono lo stesso incremento di N.O. per aggiunta di quantità fisse di TEL. La suscettività della benzina è appunto la caratteristica che fornisce una misura di tale incremento. Essa varia al variare della natura degli idrocarburi componenti. Le benzine ricche di idrocarburi paraffinici (normali e iso) hanno suscettività elevata (∆RON = 15-20), quelle ricche di aromatici hanno suscettività bassa (∆RON = 5-8). Dato che il piombo tetraetile, all’atto della combustione, potrebbe dar luogo a deposizione di piombo metallico sulla testa del cilindro e sulle sedi delle valvole, esso viene addizionato alle benzine assieme ad agenti capaci di trasformare il piombo in prodotti volatili che possono essere eliminati con lo scarico. Particolarmente usati sono il dibromoetano ed il dicloroetano che trasformano il piombo in PbBr2 o PbCl2. Una composizione tipica di agente antidetonante aggiunto alle benzine super è: TEL 63%, C2H4Br2 26%, C2H4Cl2 9%, colorante 2%. L’esigenza della eliminazione del piombo tetraetile dalle benzine, dettata da preoccupazioni circa la pericolosità del piombo per la salute dell’uomo, ha creato notevoli problemi all’industria petrolifera che ha dovuto mettere in atto provvedimenti capaci di aumentare il N.O. clear delle benzine e individuare nuovi additivi alternativi al TEL per riportare il N.O. della benzina commerciale entro i limiti richiesti per l’uso nelle autovetture. È stato possibile raggiungere questi obiettivi ricorrendo: 1) ad un incremento nella produzione di benzine di reforming che, come sarà illustrato nel seguito, sono particolarmente ricche di aromatici ed hanno quindi un N.O. clear elevato; 2) addizionando alle benzine, all’atto del blending, composti ossigenati ad alto N.O., come il metil-terz-butiletere (MTBE), l’etil-terz-butiletere (ETBE) o il terz-amilmetiletere (TAME).
Anche altri composti ossigenati, come gli alcoli, hanno elevati N.O. e potrebbero essere considerati come potenziali additivi per la formulazione di benzine senza piombo. In effetti, in ragione dell’ampia disponibilità a basso prezzo di alcool metilico (cf. processo SASOL, Cap. 3) questo alcool viene usato come additivo delle benzine in Sudafrica, così come l’alcool etilico (ottenuto per fermentazione del melasso, sottoprodotto dello zucchero di canna) è usato in Brasile. Tuttavia, gli alcoli sono molto solubili in acqua e ciò crea qualche inconveniente a livello della distribuzione del carburante e della sua combustione nel motore. Le caratteristiche principali dei diversi additivi ossigenati per benzine verdi sono riportate nella Tabella 5.
CH3 O C
CH3
CH3
CH3 CH2 O C
CH3
CH3
CH3CH3 CH3 O C
CH3
CH3
CH2 CH3
MTBE ETBE TAME
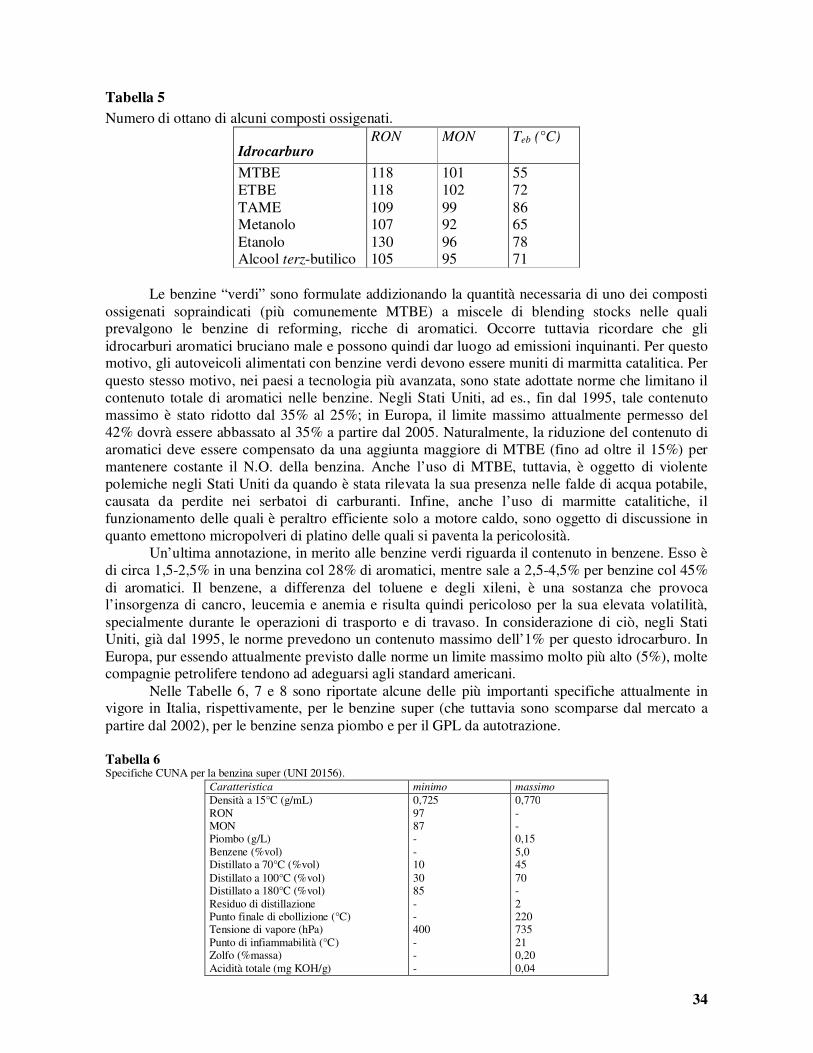
34
Tabella 5
Numero di ottano di alcuni composti ossigenati.
Idrocarburo RON MON Teb (°C)
MTBE ETBE TAME Metanolo Etanolo Alcool terz-butilico
118 118 109 107 130 105
101 102 99 92 96 95
55 72 86 65 78 71
Le benzine “verdi” sono formulate addizionando la quantità necessaria di uno dei composti ossigenati sopraindicati (più comunemente MTBE) a miscele di blending stocks nelle quali prevalgono le benzine di reforming, ricche di aromatici. Occorre tuttavia ricordare che gli idrocarburi aromatici bruciano male e possono quindi dar luogo ad emissioni inquinanti. Per questo motivo, gli autoveicoli alimentati con benzine verdi devono essere muniti di marmitta catalitica. Per questo stesso motivo, nei paesi a tecnologia più avanzata, sono state adottate norme che limitano il contenuto totale di aromatici nelle benzine. Negli Stati Uniti, ad es., fin dal 1995, tale contenuto massimo è stato ridotto dal 35% al 25%; in Europa, il limite massimo attualmente permesso del 42% dovrà essere abbassato al 35% a partire dal 2005. Naturalmente, la riduzione del contenuto di aromatici deve essere compensato da una aggiunta maggiore di MTBE (fino ad oltre il 15%) per mantenere costante il N.O. della benzina. Anche l’uso di MTBE, tuttavia, è oggetto di violente polemiche negli Stati Uniti da quando è stata rilevata la sua presenza nelle falde di acqua potabile, causata da perdite nei serbatoi di carburanti. Infine, anche l’uso di marmitte catalitiche, il funzionamento delle quali è peraltro efficiente solo a motore caldo, sono oggetto di discussione in quanto emettono micropolveri di platino delle quali si paventa la pericolosità. Un’ultima annotazione, in merito alle benzine verdi riguarda il contenuto in benzene. Esso è di circa 1,5-2,5% in una benzina col 28% di aromatici, mentre sale a 2,5-4,5% per benzine col 45% di aromatici. Il benzene, a differenza del toluene e degli xileni, è una sostanza che provoca l’insorgenza di cancro, leucemia e anemia e risulta quindi pericoloso per la sua elevata volatilità, specialmente durante le operazioni di trasporto e di travaso. In considerazione di ciò, negli Stati Uniti, già dal 1995, le norme prevedono un contenuto massimo dell’1% per questo idrocarburo. In Europa, pur essendo attualmente previsto dalle norme un limite massimo molto più alto (5%), molte compagnie petrolifere tendono ad adeguarsi agli standard americani. Nelle Tabelle 6, 7 e 8 sono riportate alcune delle più importanti specifiche attualmente in vigore in Italia, rispettivamente, per le benzine super (che tuttavia sono scomparse dal mercato a partire dal 2002), per le benzine senza piombo e per il GPL da autotrazione. Tabella 6 Specifiche CUNA per la benzina super (UNI 20156).
Caratteristica minimo massimo
Densità a 15°C (g/mL) RON MON Piombo (g/L) Benzene (%vol) Distillato a 70°C (%vol) Distillato a 100°C (%vol) Distillato a 180°C (%vol) Residuo di distillazione Punto finale di ebollizione (°C) Tensione di vapore (hPa) Punto di infiammabilità (°C) Zolfo (%massa) Acidità totale (mg KOH/g)
0,725 97 87 - - 10 30 85 - - 400 - - -
0,770 - - 0,15 5,0 45 70 - 2 220 735 21 0,20 0,04
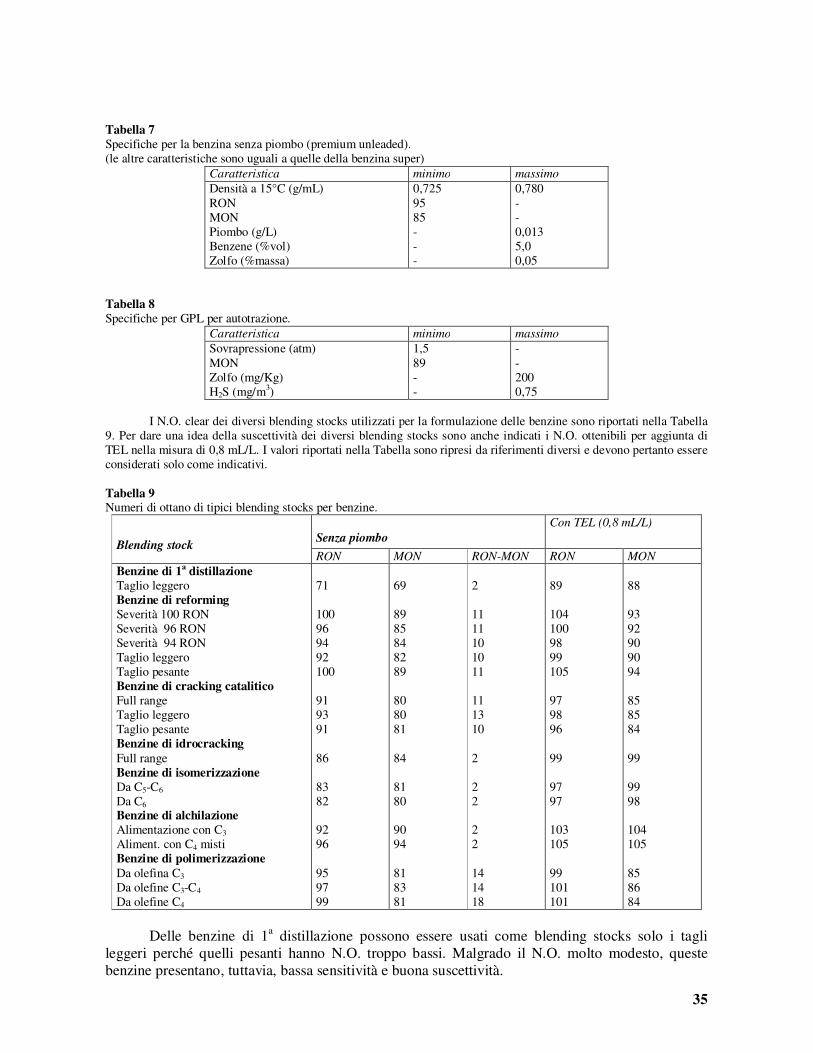
35
Tabella 7
Specifiche per la benzina senza piombo (premium unleaded). (le altre caratteristiche sono uguali a quelle della benzina super)
Caratteristica minimo massimo
Densità a 15°C (g/mL) RON MON Piombo (g/L) Benzene (%vol) Zolfo (%massa)
0,725 95 85 - - -
0,780 - - 0,013 5,0 0,05
Tabella 8
Specifiche per GPL per autotrazione. Caratteristica minimo massimo
Sovrapressione (atm) MON Zolfo (mg/Kg) H2S (mg/m3)
1,5 89 - -
- - 200 0,75
I N.O. clear dei diversi blending stocks utilizzati per la formulazione delle benzine sono riportati nella Tabella 9. Per dare una idea della suscettività dei diversi blending stocks sono anche indicati i N.O. ottenibili per aggiunta di TEL nella misura di 0,8 mL/L. I valori riportati nella Tabella sono ripresi da riferimenti diversi e devono pertanto essere considerati solo come indicativi. Tabella 9 Numeri di ottano di tipici blending stocks per benzine.
Blending stock Senza piombo
Con TEL (0,8 mL/L)
RON MON RON-MON RON MON
Benzine di 1a distillazione
Taglio leggero Benzine di reforming
Severità 100 RON Severità 96 RON Severità 94 RON Taglio leggero Taglio pesante Benzine di cracking catalitico
Full range Taglio leggero Taglio pesante Benzine di idrocracking
Full range Benzine di isomerizzazione
Da C5-C6 Da C6 Benzine di alchilazione
Alimentazione con C3 Aliment. con C4 misti Benzine di polimerizzazione
Da olefina C3 Da olefine C3-C4 Da olefine C4
71 100 96 94 92 100 91 93 91 86 83 82 92 96 95 97 99
69 89 85 84 82 89 80 80 81 84 81 80 90 94 81 83 81
2 11 11 10 10 11 11 13 10 2 2 2 2 2 14 14 18
89 104 100 98 99 105 97 98 96 99 97 97 103 105 99 101 101
88 93 92 90 90 94 85 85 84 99 99 98 104 105 85 86 84
Delle benzine di 1a distillazione possono essere usati come blending stocks solo i tagli leggeri perché quelli pesanti hanno N.O. troppo bassi. Malgrado il N.O. molto modesto, queste benzine presentano, tuttavia, bassa sensitività e buona suscettività.
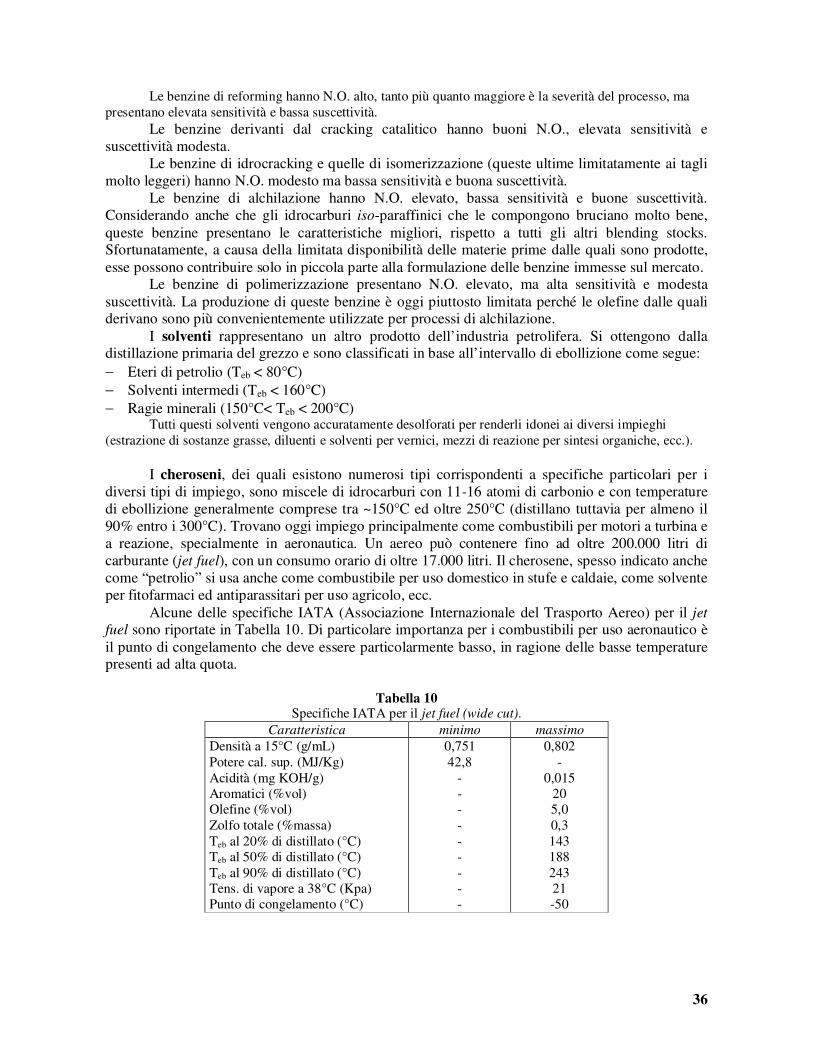
36
Le benzine di reforming hanno N.O. alto, tanto più quanto maggiore è la severità del processo, ma presentano elevata sensitività e bassa suscettività. Le benzine derivanti dal cracking catalitico hanno buoni N.O., elevata sensitività e suscettività modesta. Le benzine di idrocracking e quelle di isomerizzazione (queste ultime limitatamente ai tagli molto leggeri) hanno N.O. modesto ma bassa sensitività e buona suscettività. Le benzine di alchilazione hanno N.O. elevato, bassa sensitività e buone suscettività. Considerando anche che gli idrocarburi iso-paraffinici che le compongono bruciano molto bene, queste benzine presentano le caratteristiche migliori, rispetto a tutti gli altri blending stocks. Sfortunatamente, a causa della limitata disponibilità delle materie prime dalle quali sono prodotte, esse possono contribuire solo in piccola parte alla formulazione delle benzine immesse sul mercato. Le benzine di polimerizzazione presentano N.O. elevato, ma alta sensitività e modesta suscettività. La produzione di queste benzine è oggi piuttosto limitata perché le olefine dalle quali derivano sono più convenientemente utilizzate per processi di alchilazione. I solventi rappresentano un altro prodotto dell’industria petrolifera. Si ottengono dalla distillazione primaria del grezzo e sono classificati in base all’intervallo di ebollizione come segue: − Eteri di petrolio (Teb < 80°C) − Solventi intermedi (Teb < 160°C) − Ragie minerali (150°C< Teb < 200°C) Tutti questi solventi vengono accuratamente desolforati per renderli idonei ai diversi impieghi (estrazione di sostanze grasse, diluenti e solventi per vernici, mezzi di reazione per sintesi organiche, ecc.). I cheroseni, dei quali esistono numerosi tipi corrispondenti a specifiche particolari per i diversi tipi di impiego, sono miscele di idrocarburi con 11-16 atomi di carbonio e con temperature di ebollizione generalmente comprese tra ~150°C ed oltre 250°C (distillano tuttavia per almeno il 90% entro i 300°C). Trovano oggi impiego principalmente come combustibili per motori a turbina e a reazione, specialmente in aeronautica. Un aereo può contenere fino ad oltre 200.000 litri di carburante (jet fuel), con un consumo orario di oltre 17.000 litri. Il cherosene, spesso indicato anche come “petrolio” si usa anche come combustibile per uso domestico in stufe e caldaie, come solvente per fitofarmaci ed antiparassitari per uso agricolo, ecc. Alcune delle specifiche IATA (Associazione Internazionale del Trasporto Aereo) per il jet
fuel sono riportate in Tabella 10. Di particolare importanza per i combustibili per uso aeronautico è il punto di congelamento che deve essere particolarmente basso, in ragione delle basse temperature presenti ad alta quota.
Tabella 10
Specifiche IATA per il jet fuel (wide cut). Caratteristica minimo massimo
Densità a 15°C (g/mL) Potere cal. sup. (MJ/Kg) Acidità (mg KOH/g) Aromatici (%vol) Olefine (%vol) Zolfo totale (%massa) Teb al 20% di distillato (°C) Teb al 50% di distillato (°C) Teb al 90% di distillato (°C) Tens. di vapore a 38°C (Kpa) Punto di congelamento (°C)
0,751 42,8
- - - - - - - - -
0,802 -
0,015 20 5,0 0,3 143 188 243 21 -50
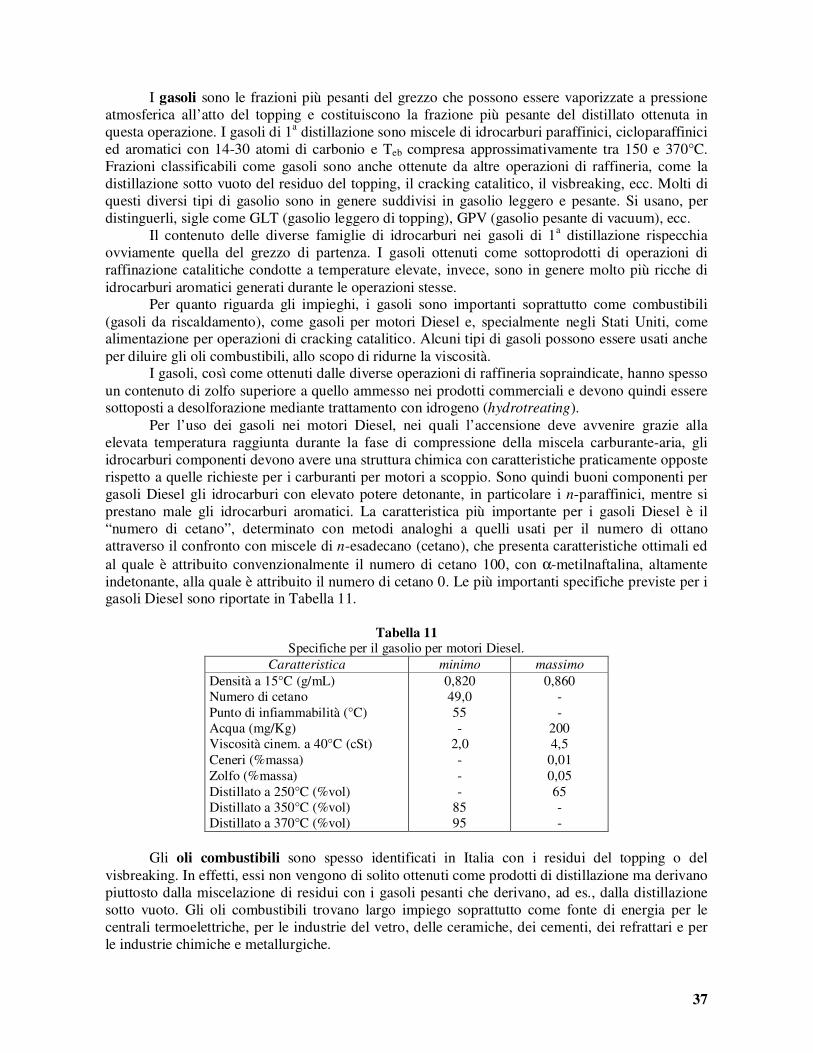
37
I gasoli sono le frazioni più pesanti del grezzo che possono essere vaporizzate a pressione atmosferica all’atto del topping e costituiscono la frazione più pesante del distillato ottenuta in questa operazione. I gasoli di 1a distillazione sono miscele di idrocarburi paraffinici, cicloparaffinici ed aromatici con 14-30 atomi di carbonio e Teb compresa approssimativamente tra 150 e 370°C. Frazioni classificabili come gasoli sono anche ottenute da altre operazioni di raffineria, come la distillazione sotto vuoto del residuo del topping, il cracking catalitico, il visbreaking, ecc. Molti di questi diversi tipi di gasolio sono in genere suddivisi in gasolio leggero e pesante. Si usano, per distinguerli, sigle come GLT (gasolio leggero di topping), GPV (gasolio pesante di vacuum), ecc.
Il contenuto delle diverse famiglie di idrocarburi nei gasoli di 1a distillazione rispecchia ovviamente quella del grezzo di partenza. I gasoli ottenuti come sottoprodotti di operazioni di raffinazione catalitiche condotte a temperature elevate, invece, sono in genere molto più ricche di idrocarburi aromatici generati durante le operazioni stesse. Per quanto riguarda gli impieghi, i gasoli sono importanti soprattutto come combustibili (gasoli da riscaldamento), come gasoli per motori Diesel e, specialmente negli Stati Uniti, come alimentazione per operazioni di cracking catalitico. Alcuni tipi di gasoli possono essere usati anche per diluire gli oli combustibili, allo scopo di ridurne la viscosità. I gasoli, così come ottenuti dalle diverse operazioni di raffineria sopraindicate, hanno spesso un contenuto di zolfo superiore a quello ammesso nei prodotti commerciali e devono quindi essere sottoposti a desolforazione mediante trattamento con idrogeno (hydrotreating). Per l’uso dei gasoli nei motori Diesel, nei quali l’accensione deve avvenire grazie alla elevata temperatura raggiunta durante la fase di compressione della miscela carburante-aria, gli idrocarburi componenti devono avere una struttura chimica con caratteristiche praticamente opposte rispetto a quelle richieste per i carburanti per motori a scoppio. Sono quindi buoni componenti per gasoli Diesel gli idrocarburi con elevato potere detonante, in particolare i n-paraffinici, mentre si prestano male gli idrocarburi aromatici. La caratteristica più importante per i gasoli Diesel è il “numero di cetano”, determinato con metodi analoghi a quelli usati per il numero di ottano attraverso il confronto con miscele di n-esadecano (cetano), che presenta caratteristiche ottimali ed al quale è attribuito convenzionalmente il numero di cetano 100, con α-metilnaftalina, altamente indetonante, alla quale è attribuito il numero di cetano 0. Le più importanti specifiche previste per i gasoli Diesel sono riportate in Tabella 11.
Tabella 11 Specifiche per il gasolio per motori Diesel.
Caratteristica minimo massimo
Densità a 15°C (g/mL) Numero di cetano Punto di infiammabilità (°C) Acqua (mg/Kg) Viscosità cinem. a 40°C (cSt) Ceneri (%massa) Zolfo (%massa) Distillato a 250°C (%vol) Distillato a 350°C (%vol) Distillato a 370°C (%vol)
0,820 49,0 55 -
2,0 - - -
85 95
0,860 - -
200 4,5
0,01 0,05 65 - -
Gli oli combustibili sono spesso identificati in Italia con i residui del topping o del visbreaking. In effetti, essi non vengono di solito ottenuti come prodotti di distillazione ma derivano piuttosto dalla miscelazione di residui con i gasoli pesanti che derivano, ad es., dalla distillazione sotto vuoto. Gli oli combustibili trovano largo impiego soprattutto come fonte di energia per le centrali termoelettriche, per le industrie del vetro, delle ceramiche, dei cementi, dei refrattari e per le industrie chimiche e metallurgiche.
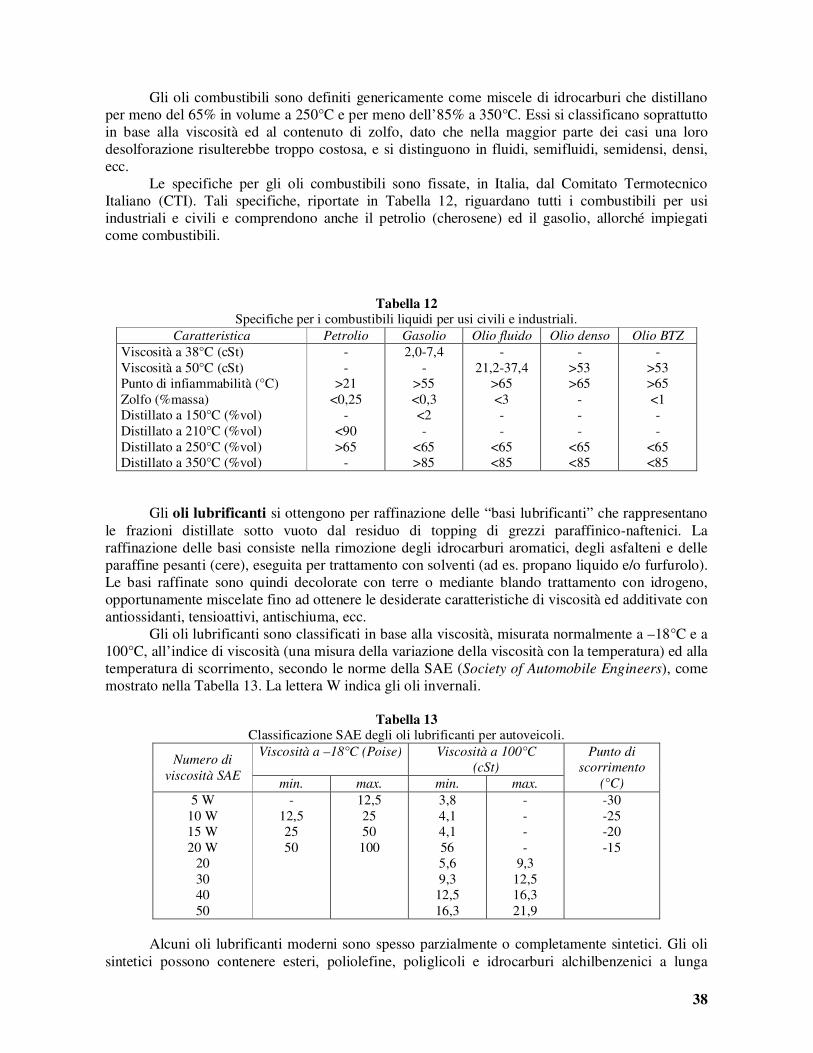
38
Gli oli combustibili sono definiti genericamente come miscele di idrocarburi che distillano per meno del 65% in volume a 250°C e per meno dell’85% a 350°C. Essi si classificano soprattutto in base alla viscosità ed al contenuto di zolfo, dato che nella maggior parte dei casi una loro desolforazione risulterebbe troppo costosa, e si distinguono in fluidi, semifluidi, semidensi, densi, ecc.
Le specifiche per gli oli combustibili sono fissate, in Italia, dal Comitato Termotecnico Italiano (CTI). Tali specifiche, riportate in Tabella 12, riguardano tutti i combustibili per usi industriali e civili e comprendono anche il petrolio (cherosene) ed il gasolio, allorché impiegati come combustibili.
Tabella 12 Specifiche per i combustibili liquidi per usi civili e industriali.
Caratteristica Petrolio Gasolio Olio fluido Olio denso Olio BTZ
Viscosità a 38°C (cSt) Viscosità a 50°C (cSt) Punto di infiammabilità (°C) Zolfo (%massa) Distillato a 150°C (%vol) Distillato a 210°C (%vol) Distillato a 250°C (%vol) Distillato a 350°C (%vol)
- -
>21 <0,25
- <90 >65
-
2,0-7,4 -
>55 <0,3 <2 -
<65 >85
- 21,2-37,4
>65 <3 - -
<65 <85
- >53 >65
- - -
<65 <85
- >53 >65 <1 - -
<65 <85
Gli oli lubrificanti si ottengono per raffinazione delle “basi lubrificanti” che rappresentano le frazioni distillate sotto vuoto dal residuo di topping di grezzi paraffinico-naftenici. La raffinazione delle basi consiste nella rimozione degli idrocarburi aromatici, degli asfalteni e delle paraffine pesanti (cere), eseguita per trattamento con solventi (ad es. propano liquido e/o furfurolo). Le basi raffinate sono quindi decolorate con terre o mediante blando trattamento con idrogeno, opportunamente miscelate fino ad ottenere le desiderate caratteristiche di viscosità ed additivate con antiossidanti, tensioattivi, antischiuma, ecc. Gli oli lubrificanti sono classificati in base alla viscosità, misurata normalmente a –18°C e a 100°C, all’indice di viscosità (una misura della variazione della viscosità con la temperatura) ed alla temperatura di scorrimento, secondo le norme della SAE (Society of Automobile Engineers), come mostrato nella Tabella 13. La lettera W indica gli oli invernali.
Tabella 13
Classificazione SAE degli oli lubrificanti per autoveicoli.
Numero di
viscosità SAE
Viscosità a –18°C (Poise) Viscosità a 100°C
(cSt)
Punto di
scorrimento
(°C) min. max. min. max.
5 W 10 W 15 W 20 W
20 30 40 50
- 12,5 25 50
12,5 25 50 100
3,8 4,1 4,1 56 5,6 9,3
12,5 16,3
- - - -
9,3 12,5 16,3 21,9
-30 -25 -20 -15
Alcuni oli lubrificanti moderni sono spesso parzialmente o completamente sintetici. Gli oli sintetici possono contenere esteri, poliolefine, poliglicoli e idrocarburi alchilbenzenici a lunga

39
catena. Gli oli multigrade contengono additivi polimerici, le macromolecole dei quali tendono ad assumere conformazioni raggomitolate a bassa temperatura mentre ad alta temperatura assumono conformazioni estese limitando così la riduzione di viscosità dovuta al riscaldamento. Ad esempio, un olio SAE-10W-40 ha un punto di scorrimento di –25°C ed una viscosità a 100°C compresa tra 12,5 e 16,3 cSt. Le paraffine sono miscele di idrocarburi n-paraffinici con almeno 20 atomi di carbonio che sono separate per cristallizzazione, con l’aiuto di adatti non-solventi, dalle basi lubrificanti. Sono solide a temperatura ambiente (cere) e sono usate soprattutto come impermeabilizzanti per carta e cartoni, come agenti sigillanti, per candele, ecc. Le vaseline ed i petrolati sono miscele di oli vegetali con isoparaffine e cicloparaffine di alto peso molecolare estratte dalle basi lubrificanti assieme alle paraffine. Le vaseline trovano applicazione per cosmetici; i petrolati come grassi lubrificanti. Grassi lubrificanti si ottengono anche miscelando oli lubrificanti con saponi metallici, argille o altre sostanze ispessenti. I bitumi di petrolio rappresentano i residui della distillazione sottovuoto o del visbreaking. Altri bitumi si ottengono anche come residuo della deasfaltazione delle basi lubrificanti con propano liquido. I bitumi contengono, secondo le norme doganali italiane, meno del 20% in massa di componenti distillabili fino a 300°C. I bitumi sono sistemi colloidali di estrema complessità. La fase dispersa (asfalteni), ricca di gruppi funzionali polari, è insolubile in idrocarburi paraffinici (propano, eptano, ecc.), ma è presente nella forma colloidale perché stabilizzata da altre sostanze (resine) che si comportano da compatibilizzanti. La fase continua (malteni), solubile in idrocarburi paraffinici, è costituita da una miscela complessa di idrocarburi pesanti con carattere più o meno aromatico A temperatura ambiente i bitumi sono solidi deformabili e fluidificano a temperature più o meno alte. La deformabilità si determina con tecniche standardizzate attraverso la misura (in decimi di mm) della profondità di penetrazione di un ago sottoposto ad un carico noto. I bitumi si classificano proprio in base al valore della penetrazione. Ad es., un bitume caratterizzato dalla sigla 80-100 è più duro (hard), o meno deformabile (soft), di un bitume 180-200. I bitumi “soffiati” sono ottenuti insufflando aria a caldo (circa 240°C) nel bitume liquido con lo scopo di ossidarlo parzialmente per migliorarne l’adesività. I bitumi “flussati” sono quelli resi fluidi a temperatura ambiente per aggiunta di cherosene o emulsionandoli con acqua. I bitumi trovano largo impiego come leganti per manti stradali (conglomerati bituminosi) e come agenti impermeabilizzanti nelle opere civili. Si usano anche come agenti impregnanti per feltri in fibra, in genere poliolefinica, da impiegare per l’impermeabilizzazione di terrazze, ecc. I bitumi possono anche essere modificati per aggiunta di piccole quantità di adatti polimeri che ne migliorano sensibilmente il comportamento reologico e le proprietà meccaniche a temperatura ambiente, rendendoli adatti alla produzione di conglomerati bituminosi drenanti e fonoassorbenti.
______________________


















