Biologia del canale gastrointestinale · la differenziazione, la senescenza, il ciclo cellulare e...
15
SEZIONE I Biologia del canale gastrointestinale Capitolo 1 Crescita cellulare e neoplasia .................................................................. 3 Capitolo 2 Immunità mucosale e infiammazione ..................................................... 16 Capitolo 3 Microbiota intestinale ............................................................................ 28 Capitolo 4 Ormoni e neurotrasmettitori gastrointestinali .......................................... 37
Transcript of Biologia del canale gastrointestinale · la differenziazione, la senescenza, il ciclo cellulare e...

SEZIONE
I
Biologia del canale gastrointestinaleCapitolo 1 Crescita cellulare e neoplasia .................................................................. 3
Capitolo 2 Immunità mucosale e infiammazione ..................................................... 16
Capitolo 3 Microbiota intestinale ............................................................................ 28
Capitolo 4 Ormoni e neurotrasmettitori gastrointestinali .......................................... 37
001_SLEISENGER_01_0003_0015.indd 1 24/02/18 15:44

001_SLEISENGER_01_0003_0015.indd 2 24/02/18 15:44

Capitolo 1 Crescita cellulare e neoplasia 3
3
INDICE CAPITOLOMeccanismi della normale omeostasi cellulare .................................. 3
Proliferazione cellulare ........................................................................3Apoptosi .............................................................................................4Senescenza ........................................................................................4Vie di segnalazione che regolano la crescita cellulare ...........................4
Sviluppo dei tumori intestinali ........................................................... 6Carcinogenesi sequenziale multistep ....................................................6Espansione clonale .............................................................................8Cellule staminali tumorali ....................................................................8
Geni associati alle neoplasie ............................................................. 8Oncogeni ............................................................................................8Geni oncosoppressori ..........................................................................9Geni deputati alla riparazione del DNA ...............................................10Vie di segnalazione degli oncogeni ....................................................11RNA non codificanti ..........................................................................11Epigenetica .......................................................................................12
Crescita cellulare e neoplasia
CAPITOLO
1 MANISH K. GALA E DANIEL C. CHUNG
Metabolismo del tumore .................................................................. 12Influenze ambientali e microambientali ........................................... 12
Carcinogenesi chimica ......................................................................12Fattori dietetici ..................................................................................12Microbioma ......................................................................................13Cancro e infiammazione ....................................................................13
Aspetti biologici delle metastasi tumorali......................................... 13Transizione epitelio-mesenchima .......................................................13Angiogenesi e linfoangiogenesi ..........................................................14
Medicina molecolare: approcci attuali e futuri all’oncologia gastrointestinale .......................................................................... 14
Diagnostica molecolare .....................................................................14Studi di genome-wide association .....................................................14Sequenziamento dell’intero genoma e dell’esoma ..............................15
Le neoplasie del tratto gastrointestinale (GI) sono tra le più fre-quenti patologie che giungono all’osservazione dei gastroentero-logi. I notevoli progressi nella comprensione delle basi cellulari e molecolari di queste neoplasie hanno fornito le basi per lo sviluppo di nuovi approcci preventivi, diagnostici e terapeutici. Nonostante alcune caratteristiche della carcinogenesi siano spe-cifiche dei diversi tessuti, molti meccanismi sono comuni in tutti i distretti del tratto GI. Questo capitolo esamina i meccanismi della crescita cellulare, sia normale sia patologica, e le alterazioni molecolari che facilitano la trasformazione maligna. I concetti di base discussi in questo capitolo forniscono le nozioni generali per la discussione delle specifiche neoplasie GI nei capitoli successivi.
MECCANISMI DELLA NORMALE OMEOSTASI CELLULARE
Proliferazione cellulareLa trasformazione neoplastica deriva dalla perturbazione di un’in-tricata rete di meccanismi omeostatici che regolano la progressione, la differenziazione, la senescenza, il ciclo cellulare e la morte cellulare programmata. La proliferazione si verifica quando la cellula attraversa il ciclo cellulare (Fig. 1-1). In preparazione alla divisione cellulare, vi è un periodo di attività biosintetica chia-mato fase G1 che è tipicamente associato a un incremento delle dimensioni cellulari. Questa fase è seguita da una precisa dupli-cazione del genoma, definita fase S. Dopo un periodo intermedio, definito fase G2, si verifica la mitosi nella fase M.
L’impegno a procedere alla replicazione del DNA si verifica dalla fase G1 con il superamento del checkpoint G1/S definito punto di restrizione (R). Le cellule possono uscire da questa fase di attiva
proliferazione prima di raggiungere il punto R ed entrare in una fase quiescente, G0. Le cellule possono successivamente rientrare nel ciclo cellulare uscendo dallo stato G0 (vedi Fig. 1-1). Esiste un altro checkpoint al confine tra la fase G2 e la M. Il checkpoint G2/M assicura che la mitosi non proceda prima della riparazione di qualsiasi danno al DNA dopo la replicazione del genoma. Una compromissione della funzione di questi checkpoint si osserva frequentemente nel cancro.
La regolazione della progressione del ciclo cellulare è realiz-zata principalmente dall’attività delle cicline e dalle chinasi cicli-na-dipendenti che operano a livello dei checkpoint G1/S e G2/M. Le cicline A e B sono prevalentemente espresse durante le fasi S e G2, (vedi Fig. 1-1). Al contrario, le cicline D ed E sono maggior-mente attive durante la fase G1.1 L’aumentata espressione della ciclina D1 nei fibroblasti risulta in un più rapido ingresso delle cellule nella fase S. L’espressione della ciclina D1 è significativa-mente aumentata in molte neoplasie, incluse quelle del tratto GI.2
Ogni ciclina forma un complesso con una chinasi ciclina- dipendente (CDK) in un determinato momento del ciclo cellulare. Le cicline funzionano da catalizzatori per l’attività delle CDK (vedi Fig. 1-1). I complessi ciclina-CDK regolano la progressione del ciclo cellulare attraverso la fosforilazione di proteine chiave quali il prodotto del gene del retinoblastoma (pRb) e alcuni mem-bri della sua famiglia, come p130 e p107.3 Il risultato finale è la progressione dalla fase G1 alla fase S del ciclo cellulare.
Il ciclo cellulare è anche regolato da molteplici inibitori delle CDK; tra questi, p21CIP1/WAF1 e p27KIP1 sono inibitori della ciclina E/CDK2. Originariamente scoperto come facente parte del complesso contenente la ciclina D1 e CDK4/6, p21CIP1/WAF1 è trascrizionalmente attivato da diversi geni oncosoppressori, in particolare TP53.4 Un altro inibitore delle CDK, p16INK4A, inibi-sce specificamente CDK4 e CDK6 ed è parte di una più ampia famiglia di inibitori correlati, che include p14, p15 e p18;5 p16INK4A
001_SLEISENGER_01_0003_0015.indd 3 24/02/18 15:44

4 Sezione I Biologia del canale gastrointestinale
è frequentemente inattivato nella maggior parte dei tumori GI, in accordo con la sua funzione di gene oncosoppressore.6,7 È noto che p16INK4A interrompe il complesso tra la ciclina D1 e CDK4/6, liberando in tal modo p21CIP1/WAF1 e p27KIP1 e inibendo così l’atti-vità della ciclina E/CDK2.8 In aggiunta, l’espressione di p16INK4A determina un aumento della stabilità dell’oncosoppressore p53.9
ApoptosiL’apoptosi (morte cellulare programmata) è un importante meccanismo che controbilancia la proliferazione cellulare e l’e-vasione dal normale meccanismo apoptotico riveste un ruolo cri-tico nell’oncogenesi. L’apoptosi è caratterizzata da eventi correlati che includono il compattamento della cromatina, la condensa-zione del citoplasma e la conseguente picnosi nucleare. Questi
cambiamenti sono seguiti dalla frammentazione nucleare con formazione di corpi apoptotici, che rappresentano il residuo cellulare che in seguito viene eliminato attraverso la fagocitosi da parte della componente cellulare macrofagica.
L’apoptosi può essere innescata da stimoli interni o esterni. Abitualmente, l’apoptosi si verifica durante il normale sviluppo per facilitare la differenziazione tissutale. Stimoli interni che pos-sono innescare il processo apoptotico includono la deprivazione di nutrienti, l’ipossia, il danno al DNA e lo stress ossidativo. Alla fine, questi segnali apoptotici interni convergono per incremen-tare la permeabilità della membrana mitocondriale, con conse-guente perdita del gradiente elettrico necessario per la respira-zione aerobia (Fig. 1-2). I piccoli attivatori delle caspasi derivati dai mitocondri (SMACs) e il citocromo c sono rilasciati nel cito-plasma. SMACs e il cosiddetto complesso apoptosoma (citocromo c, caspasi 9 e Apaf1) attivano dunque le caspasi, come la caspasi 3, accelerando la morte cellulare. Le caspasi sono cisteina-pro-teasi intracellulari e costituiscono mediatori chiave della morte cellulare programmata nelle cellule eucariotiche.
È stato dimostrato che le proteine della famiglia di Bcl-2 modulano l’attività dei pori e la permeabilità mitocondriale. Bax e Bak promuovono la formazione dei pori, mentre Bcl-2, Bcl-xL e McL-1 la inibiscono. Il rapporto stechiometrico tra mem-bri proapoptotici e antiapoptotici di questa famiglia determina l’equilibrio tra sopravvivenza e morte cellulare.10 Nelle neoplasie, questo equilibrio è sbilanciato verso i fattori antiapoptotici.
L’apoptosi può anche essere stimolata da segnali esterni. L’attivazione, per esempio, dei recettori TNF, TNFR1 e TNFR2 deter-mina l’attivazione delle caspasi. L’attivazione del recettore Fas e di FAS ligando esita, a sua volta, nella cascata di segnalazione che porta all’attivazione delle caspasi. In aggiunta a queste vie ben caratteriz-zate, altri fattori come tossine, segnali chimici e patogeni di varia natura possono innescare il processo apoptotico (vedi Fig. 1-2).
SenescenzaLa senescenza è il processo attraverso cui le cellule perdono defi-nitivamente la loro capacità di dividersi. La senescenza può veri-ficarsi in risposta allo stress indotto dall’attivazione di oncogeni, di un danno al DNA o dopo un numero fisso di divisioni cellulari (senescenza replicativa). Questi processi limitano la proliferazione sregolata o eccessiva, ma contribuiscono anche all’invecchiamento e all’esaurimento di cellule staminali.11 Durante la carcinogenesi, questi meccanismi oncosoppressori vengono aggirati o sono com-pletamente persi.
Quando cresce in vitro, la maggior parte delle cellule ha un potenziale replicativo limitato e, alla fine, va incontro a senescenza replicativa.12 I telomeri sono sequenze ripetitive di DNA poste alle estremità di tutti i cromosomi, che regolano la stabilità cromoso-mica. I telomeri si accorciano dopo ciascuna divisione cellulare e quando raggiungono una certa lunghezza critica si innesca il processo di senescenza, mediante attivazione di segnali che por-tano al danno del DNA. Le cellule cancerose hanno la capacità di mantenere la lunghezza dei loro telomeri nonostante molteplici divisioni cellulari attraverso la riattivazione dell’attività dell’en-zima telomerasi, che aggiunge telomeri addizionali alle estremità dei cromosomi.13 Una segnalazione aberrante di danno del DNA può determinare, nel cancro, fusioni cromosomiche e aneuploidia quando i telomeri si esauriscono.
Vie di segnalazione che regolano la crescita cellulareLa proliferazione cellulare si attua attraverso l’uscita della cel-lula dalla fase di arresto G0 del ciclo cellulare (vedi Fig. 1-1). Anche se la progressione attraverso il ciclo cellulare è control-lata dai meccanismi regolatori descritti precedentemente, nel suo complesso la proliferazione è modulata anche da stimoli esterni. I fattori di crescita che legano specifici recettori transmembrana
Regolazione del ciclo cel lulare mediante cicl in(cycs), chinasi ciclino-dipendenti (cdks) e cdk-inibitori. Nel normale ciclo cellulare, la sintesi del DNA (in cui il DNA cromosomico viene duplicato) si verifica nella fase S, mentre la mitosi (in cui prima i nuclei si dividono per formare una coppia di nuovi nuclei, con successiva divisione cellulare per formare una coppia di cellule figlie) avviene durante la fase M. La fase S e la fase M sono separate da altre due fasi, la fase G1, dopo la mitosi e prima della sintesi del DNA, e la fase G2, che segue la fase S. Durante queste fasi, la cellula sintetizza proteine e meta-boliti, incrementando la sua massa e preparandosi per la fase S e la fase M. La progressione del ciclo cellulare è regolata in primo luogo in due punti, i checkpoint G2/M e G1/S, attraverso le attività coordinate di cicline e CDK, che a loro volta sono regolate negativa-mente dai CDK-inibitori (famiglie INK4 e CIP/KIP). La fase G1 inter-media è caratterizzata dall’interazione tra ciclina D1 e cdk4/6. Questo complesso iperfosforila la proteina del retinoblastoma (pRb) e i mem-bri della sua famiglia (per es., p130). Un altro importante complesso che si forma tra le fasi G1 e S è quello tra cdk2 e ciclina E (cyc E). Il risultato è il rilascio di fattori della trascrizione come E2F, che sono complessati con pRb. A sua volta, E2F lega e attiva i promotori di geni importanti nella sintesi del DNA.
o p21CIP1/WAF1
cdk2
cyc E
cdk4/6
cyc D1
E2FG1
PP
P
GO
M
G2
S
Ciclina B
Ciclina A
p27KIP1
–– +
pRbpRb
p16INK4A
E2F
001_SLEISENGER_01_0003_0015.indd 4 24/02/18 15:44

Capitolo 1 Crescita cellulare e neoplasia 5
sulla superficie cellulare hanno un ruolo centrale nella regolazione della proliferazione. Le code citoplasmatiche di queste proteine recettoriali transmembrana attivano cascate di segnali intracellulari, in seguito al legame con il proprio ligando. In aggiunta a fattori di crescita peptidici, la matrice extracellulare e le molecole di adesione cellula-cellula (integrine, caderine, selectine, proteoglicani) pos-sono avere un impatto significativo sulla proliferazione cellulare. Alterazioni della matrice cellulare o delle interazioni cellula-cel-lula sono particolarmente importanti nel contribuire al fenotipo invasivo delle cellule maligne.
L’interazione di ligandi con i loro recettori sulla superficie cellulare induce segnali intracellulari che alterano la trascrizione genica e l’espressione proteica. Sembra che tre importanti sottotipi recettoriali inizino la segnalazione cellulare attraverso l’intera-zione ligando-recettore sulla superficie cellulare: (1) tirosinchinasi; (2) serin- e treoninchinasi; (3) recettori accoppiati a proteine G.
I recettori per alcuni fattori di crescita peptidici contengono attività tirosinchinasica intrinseca all’interno della loro coda intracellulare. A seguito del legame con il ligando, l’attività
tirosinchinasica viene stimolata, determinando la fosforilazione dei residui di tirosina di proteine target all’interno della cellula. La maggior parte dei recettori determina un’autofosforilazione dei residui tirosinici presenti nei recettori stessi per amplificare il segnale e, in alcuni casi, si ha una conseguente attenuazione della loro stessa attività, determinando un feedback regolatorio intramolecolare. I recettori per diversi fattori di crescita peptidici, tra cui EGF, appartengono a questa classe recettoriale.
Altri recettori della superficie cellulare possiedono attività chinasica verso i residui di serina o treonina, piuttosto che quelli tirosinici. Molteplici siti di fosforilazione di serina e treonina sono presenti su diversi recettori per fattori di crescita, inclusi i recettori tirosinchinasici, suggerendo l’esistenza di significative interazioni nel contesto di vari recettori presenti su una singola cellula.14 Il complesso recettoriale del fattore di crescita TGF-β è un impor-tante esempio di recettore transmembrana contenente attività chinasica verso i residui di serina e treonina.
Molti recettori sono membri della cosiddetta famiglia dei recettori a 7 domini transmembrana. Questi recettori sono accoppiati
FIGURA 1-1. L’apoptosi (morte cellulare programmata) controbilancia la proliferazione cellulare per regolare la crescita tissutale complessiva. La complessa interazione tra molecole proapoptotiche e antiapoptotiche risulta in un’attivazione a cascata di caspasi che mediano la morte cellulare. Alcuni di questi segnali vengono innescati attraverso insulti ambientali che attivano l’oncosoppressore tumorale TP53, alcuni mediante recettori di morte cellulare, quali TNF-R1, TNF-R2eFas. In aggiunta, esiste un’interazione tra molecole proapoptotiche (Bax, Bak) e antiapoptotiche (BCL-2, BCL-XL). Entrambe le vie convergono sui mitocondri, portando al rilascio del citocromo c e alla formazione del complesso apoptosoma (APAF1, caspasi 9 e citocromo c). Questo conduce all’attivazione di molteplici caspasi, a un danneggiamento del DNA e infine alla morte cellulare. BID, dominio di interazione bcl-2.
Caspasi
Morte cellulare
Recettore del TNFo Fas
Citocromo c
Complessoapoptosoma
Mitocondrio
Caspasi 9
Danno del DNA
001_SLEISENGER_01_0003_0015.indd 5 24/02/18 15:44

6 Sezione I Biologia del canale gastrointestinale
a proteine che legano nucleotidi guaninici e sono definite proteine G. Le proteine G subiscono un cambiamento conformazionale che è dipendente dalla presenza di guanosinfosfati.15 L’attivazione delle proteine G può innescare una varietà di segnali intracellulari, tra cui la stimolazione della fosfolipasi C e la generazione di fosfatidil-inositolo (principalmente inositolo 1,4,5-trifosfato) e diacilglicerolo attraverso l’idrolisi dei fosfolipidi di membrana, così come la modulazione di secondi messaggeri quali cAMP e GMP.16 I recettori della somatostatina sono un esempio di recettore accoppiato a proteine G prevalente nel tratto GI.
Il legame di fattori di crescita e citochine agli specifici recettori della superficie cellulare produce alterazioni in una varietà di funzioni cellulari che influenza un’ampia gamma di meccanismi omeostatici che regola la crescita cellulare. Queste funzioni inclu-dono l’attivazione dei trasportatori ionici, l’assorbimento di nutrienti e l’incremento della sintesi proteica.
La via Wnt è un importante esempio di via di segnalazione che regola numerosi meccanismi omeostatici per il controllo della proliferazione delle cellule dell’epitelio intestinale (Fig. 1-3). Questa via è conservata nel corso dell’evoluzione tra le diverse specie ed è essenziale per l’accumulo di β-catenina nel nucleo, dove legandosi al fattore trascrizionale Tcf-4 modula una serie di geni target coinvolti nella proliferazione.17 Nelle cellule normali questo segnale è innescato da specifici ligandi di Wnt
che si legano ai recettori della famiglia Frizzled localizzati sulla superficie cellulare. L’inibizione del segnale Wnt nei topi può essere ottenuta mediante la delezione di Tcf-4 o l’espressione dell’inibitore di Wnt Dickkopf1, con conseguente marcata ridu-zione della proliferazione dell’epitelio intestinale.18-19 L’omeostasi tissutale è mantenuta anche da segnali inibitori della crescita che controbilanciano quelli proliferativi; per esempio, il TGF-β non solo induce la trascrizione degli inibitori del ciclo cellulare p15INK4B e p21CIP1/WAF1, ma incrementa anche l’attività inibitoria di p27KIP1 sul complesso ciclina E/CDK2 (vedi Fig. 1-1).20 Questi effetti del TGF-β sono mediati a livello intracellulare attraverso le proteine della famiglia Smad.
SVILUPPO DEI TUMORI INTESTINALI
Carcinogenesi sequenziale multistepMolteplici alterazioni genetiche sequenziali sono necessarie per la trasformazione del normale epitelio intestinale in neoplasia. Questa natura multistep della carcinogenesi è stata ampiamente caratterizzata nello sviluppo del cancro del colon (vedi Cap. 127). L’accumulo di alterazioni genetiche ed epigenetiche mostra un parallelismo con la progressione da normale epitelio attraverso
FIGURA 1-2. Il pathway di segnalazione Wnt è un importante regolatore della proliferazione dell’epitelio intestinale e della tumorigenesi. In assenza del segnale Wnt (in alto a sinistra), la β-catenina citosolica forma un complesso citoplasmatico con APC, axina e glicogeno sintasi chinasi-3β (GSK-3β). Questo complesso fosforila la β-catenina stessa e la indirizza verso la degradazione attraverso la via proteasomica di ubiquitinazione. In presenza di un segnale Wnt attivo (in alto a destra), la β-catenina è stabilizzata e la quantità citoplasmatica in eccesso viene traslocata nel nucleo, dove interagisce con il fattore di trascrizione Tcf-4 per regolare l’espressione di alcuni geni chiave. APC, poliposi adenomatosa del colon; P, gruppo fosfato; VEGF, fattore di crescita dell’endotelio vascolare.
Recettore frizzled Membrana plasmatica
Dishevelled
Proteosoma
NucleoCiclina D1
b-cateninacitosolica
AxinaAxina
-catenina
001_SLEISENGER_01_0003_0015.indd 6 24/02/18 15:44
Capitolo 1 Crescita cellulare e neoplasia 7
polipi adenomatosi fino a neoplasia maligna. Studi di patogenesi molecolare del cancro del colon sono serviti come paradigma per chiarire le alterazioni genetiche di altri tumori del tratto GI, com-presi i tumori maligni dello stomaco e del pancreas.
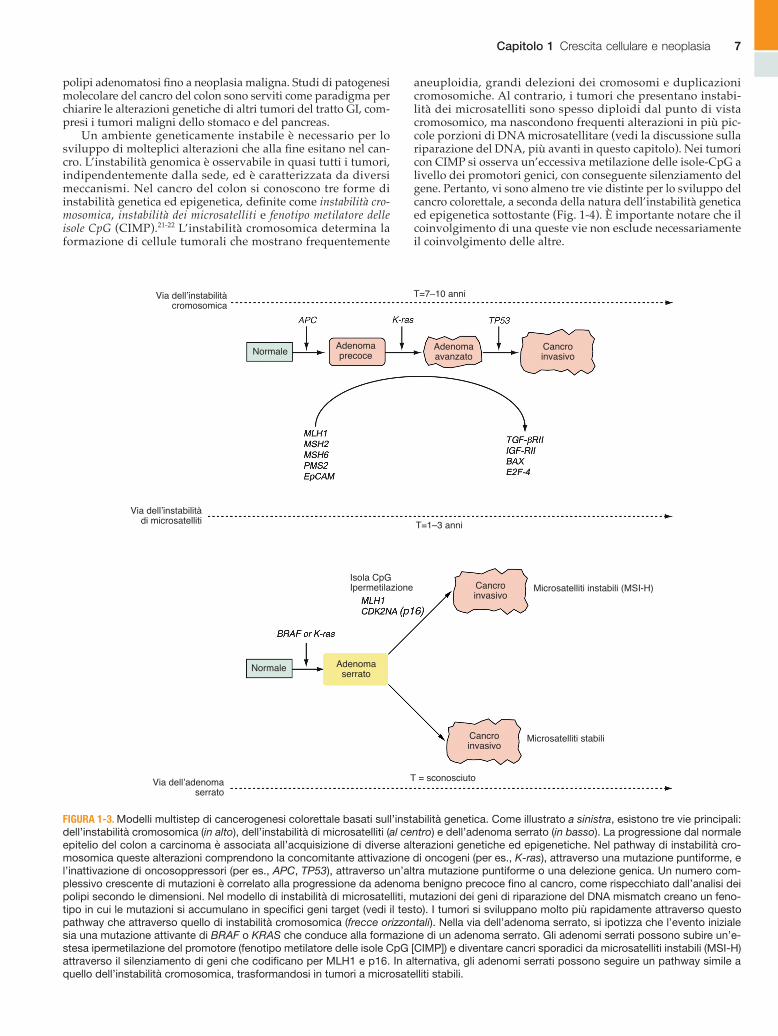
Un ambiente geneticamente instabile è necessario per lo sviluppo di molteplici alterazioni che alla fine esitano nel can-cro. L’instabilità genomica è osservabile in quasi tutti i tumori, indipendentemente dalla sede, ed è caratterizzata da diversi meccanismi. Nel cancro del colon si conoscono tre forme di instabilità genetica ed epigenetica, definite come instabilità cro-mosomica, instabilità dei microsatelliti e fenotipo metilatore delle isole CpG (CIMP).21-22 L’instabilità cromosomica determina la formazione di cellule tumorali che mostrano frequentemente
aneuploidia, grandi delezioni dei cromosomi e duplicazioni cromosomiche. Al contrario, i tumori che presentano instabi-lità dei microsatelliti sono spesso diploidi dal punto di vista cromosomico, ma nascondono frequenti alterazioni in più pic-cole porzioni di DNA microsatellitare (vedi la discussione sulla riparazione del DNA, più avanti in questo capitolo). Nei tumori con CIMP si osserva un’eccessiva metilazione delle isole-CpG a livello dei promotori genici, con conseguente silenziamento del gene. Pertanto, vi sono almeno tre vie distinte per lo sviluppo del cancro colorettale, a seconda della natura dell’instabilità genetica ed epigenetica sottostante (Fig. 1-4). È importante notare che il coinvolgimento di una queste vie non esclude necessariamente il coinvolgimento delle altre.
FIGURA 1-3. Modelli multistep di cancerogenesi colorettale basati sull’instabilità genetica. Come illustrato a sinistra, esistono tre vie principali: dell’instabilità cromosomica (in alto), dell’instabilità di microsatelliti (al centro) e dell’adenoma serrato (in basso). La progressione dal normale epitelio del colon a carcinoma è associata all’acquisizione di diverse alterazioni genetiche ed epigenetiche. Nel pathway di instabilità cro-mosomica queste alterazioni comprendono la concomitante attivazione di oncogeni (per es., K-ras), attraverso una mutazione puntiforme, e l’inattivazione di oncosoppressori (per es., APC, TP53), attraverso un’altra mutazione puntiforme o una delezione genica. Un numero com-plessivo crescente di mutazioni è correlato alla progressione da adenoma benigno precoce fino al cancro, come rispecchiato dall’analisi dei polipi secondo le dimensioni. Nel modello di instabilità di microsatelliti, mutazioni dei geni di riparazione del DNA mismatch creano un feno-tipo in cui le mutazioni si accumulano in specifici geni target (vedi il testo). I tumori si sviluppano molto più rapidamente attraverso questo pathway che attraverso quello di instabilità cromosomica (frecce orizzontali). Nella via dell’adenoma serrato, si ipotizza che l’evento iniziale sia una mutazione attivante di BRAF o KRAS che conduce alla formazione di un adenoma serrato. Gli adenomi serrati possono subire un’e-stesa ipermetilazione del promotore (fenotipo metilatore delle isole CpG [CIMP]) e diventare cancri sporadici da microsatelliti instabili (MSI-H) attraverso il silenziamento di geni che codificano per MLH1 e p16. In alternativa, gli adenomi serrati possono seguire un pathway simile a quello dell’instabilità cromosomica, trasformandosi in tumori a microsatelliti stabili.
Via dell’instabilitàcromosomica
Normale
T=7–10 anni
Adenomaprecoce
Via dell’instabilitàdi microsatelliti
Normale
Adenomaavanzato
Cancroinvasivo
T=1–3 anni
Isola CpGIpermetilazione Cancro
invasivo
Cancroinvasivo
T = sconosciuto
Adenomaserrato
Microsatelliti instabili (MSI-H)
Microsatelliti stabili
Via dell’adenomaserrato
001_SLEISENGER_01_0003_0015.indd 7 24/02/18 15:44

8 Sezione I Biologia del canale gastrointestinale
Espansione clonaleL’espansione clonale è essenziale per lo sviluppo del tumore.23 Mentre le mutazioni germinali possono condurre a un’alterata espressione di un gene in tutte le cellule di un tessuto, le mutazioni somatiche generalmente si verificano solo in una piccola sottopo-polazione di cellule. L’espansione clonale di queste cellule mutate si verifica se una mutazione genica specifica comporta un vantag-gio di sopravvivenza per le cellule. Una seconda fase di espan-sione clonale ha luogo quando una cellula all’interno di questa popolazione subisce ancora un’altra alterazione genetica che ne incrementa ulteriormente le proprietà di crescita. Questo processo di selezione, con l’accumulo di alterazioni genetiche, determina la trasformazione cellulare e l’acquisizione di un fenotipo maligno. Una volta che una franca malignità si è sviluppata, l’elenco delle mutazioni albergate può variare tra le cellule cancerose. Questo aspetto è definito eterogeneità tumorale e può conferire una serie di vantaggi nella selezione cellulare.24 Un esempio tipico sono le metastasi tumorali, che possono essere favorite dalla selezione di un sottogruppo di cellule che acquisiscono la capacità di attra-versare il sistema circolatorio e prosperare in un nuovo ambiente tissutale lontano dal tessuto primitivo.
Cellule staminali tumoraliQueste osservazioni sull’eterogeneità tumorale hanno portato all’ipotesi delle cellule staminali tumorali, che suggerisce l’esi-stenza di un sottogruppo di cellule tumorali che presenta pro-prietà simili a quelle delle cellule staminali (Cancer Stem Cells, CSCs). Molti studi hanno portato all’ipotesi che le CSCs siano cellule che innescano il tumore, da cui parte l’espansione clonale. In aggiunta, è stato ipotizzato che l’eradicazione di queste cellule possa costituire un obiettivo terapeutico chiave, in quanto esse sembrano responsabili delle recidive tumorali e della resistenza al trattamento chemioterapico. Due diverse ipotesi sul ruolo delle CSCs sono state recentemente formulate.25 La prima individua un modello gerarchico in cui le CSCs sarebbero le progenitrici delle cellule tumorali, con un potenziale riproduttivo limitato. Il secondo modello ipotizza che ciascuna cellula tumorale possa potenzialmente comportarsi come una CSC, ma che questa deter-minazione sia stocasticamente basata su fattori intrinseci alla cel-lula, in aggiunta a segnali ambientali esterni che determinano la spinta in senso staminale. L’analisi delle CSCs ha dimostrato la presenza di programmi trascrizionali e marker condivisi con le normali cellule staminali intestinali. Marker come Lgr5 e EphB2, tipici delle cellule staminali dell’epitelio intestinale, sono stati usati per identificare e purificare CSCs del carcinoma del colon.26
GENI ASSOCIATI ALLE NEOPLASIEI geni che complessivamente rivestono un ruolo importante nell’oncogenesi generalmente conducono a un sovvertimento degli ordinati meccanismi della normale proliferazione cellulare. Dal momento che la normale proliferazione cellulare dipende da un’ampia varietà di geni, non sorprende che alterazioni nell’e-spressione di un certo gruppo di geni siano responsabili, in parte o completamente, delle caratteristiche fenotipiche di trasforma-zione. Nonostante tale diversità, tutti questi geni alterati sembrano appartenere a uno dei seguenti due gruppi distinti: (1) oncogeni, che conferiscono attivamente proprietà promuoventi la crescita; (2) geni oncosoppressori, i cui prodotti normalmente bloccano la crescita o la proliferazione. Un’importante categoria all’interno dei geni oncosoppressori include i geni della riparazione del DNA, che prevengono l’accumulo di nuove mutazioni. L’attivazione di oncogeni o l’inattivazione di geni oncosoppressori contribui- sce alla trasformazione maligna. Siti trascrizionalmente attivi del genoma che non codificano per proteine rivestono anch’essi un ruolo significativo nella regolazione dell’espressione genica
e della carcinogenesi. Questi RNA non codificanti possono svol-gere, allo stesso tempo, funzioni oncogeniche e oncosoppressive.
OncogeniTipicamente, gli oncogeni sono geni che codificano per una proteina cellulare normale espressa a livelli inappropriati o geni mutati che producono una proteina strutturalmente alterata che esercita un’attività anomala. Per esempio, diversi geni che codificano per recettori di fattori di crescita contenenti tirosinchinasi diventano oncogeni dopo una mutazione che determina un’attività tirosin-chinasica incontrollata, che non è più dipendente dalla presenza dell’appropriato ligando. I geni cellulari normali da cui gli onco-geni derivano sono definiti proto-oncogeni. La maggior parte di questi geni è espressa in vari tipi di cellule tumorali.
Molteplici meccanismi possono determinare l’attivazione di un oncogene. Tra questi vi sono la trasduzione o inserzione genica, la mutazione puntiforme, il riarrangiamento genico e l’amplifica-zione genica. La trasduzione e l’inserzione genica solitamente sono il risultato di un’infezione retrovirale. Le mutazioni puntiformi possono determinare l’espressione di oncogeni costitutivamente attivi. I riarrangiamenti genici possono condurre alla formazione di proteine di fusione oncogene e l’amplificazione genica conduce a una sovraespressione incontrollata di un normale prodotto genico.
Le proteine codificate dagli oncogeni comprendono almeno quattro gruppi distinti: fattori di crescita peptidici che possono essere secreti all’interno del milieu extracellulare, proteinchinasi, proteine che trasducono il segnale associate alla superficie interna della membrana cellulare (proteine G membrana-associate) e proteine regolatrici della trascrizione localizzate nel nucleo.
Oncogeni correlati fattori di crescita peptidiciGli effetti trasformanti dell’espressione amplificata di fattori di crescita è stata dimostrata sia in vitro sia in vivo. Diverse proteine correlate a fattori di crescita codificate dagli oncogeni sono state individuate, compresa la famiglia delle proteine Wnt e Sis, che codifica per la catena β del fattore di crescita derivato dalle pia-strine. Le cellule cancerose possono innescare segnali autocrini per promuovere la propria crescita o modulare segnali, in modo che lo stroma adiacente secerna quantità elevate di fattori di crescita per l’epitelio trasformato.
Oncogeni correlati a proteinchinasiLa più grande famiglia di oncogeni codifica per proteine con attivi- tà proteinchinasica. Questi oncogeni comprendono l’intera varietà di proteinchinasi, incluse le tirosinchinasi recettoriali/non recettoriali e le serin-/treoninchinasi citoplasmatiche. Alcuni membri di questo ampio gruppo di oncogeni sono espressi da neo-plasie del tratto GI, tra cui il recettore tirosinchinasico della famiglia di recettori EGF (ERBB1-4) e la tirosinchinasi non recettoriale Src associata alla superficie interna della membrana plasmatica.
Oncogeni correlati a segnali di trasduzione (proteine G membrana-associate)I passaggi intermedi che traducono efficientemente il legame ligan-do-recettore in un segnale intracellulare sono essenziali nel mediare le risposte funzionali della cellula. Mutazioni dei geni che codi-ficano per proteine chiave che partecipano alla trasduzione del segnale possono condurre a trasformazione cellulare.
Le proteine G regolano il segnale di un’ampia famiglia di recettori accoppiati a proteine G (GPCRs) attraverso lo scambio del guanosintrifosfato (GTP) con il guanosindifosfato (GDP). I geni ras codificano per una famiglia di proteine correlata alle proteine G e sono tra gli oncogeni più comunemente riscontrati nei tumori maligni dell’apparato GI. La famiglia ras contiene 3 geni: H-ras, K-ras e N-ras. Mutazioni puntiformi che determinano sostituzioni aminoacidiche in posizioni critiche convertono il gene normale in un oncogene.
001_SLEISENGER_01_0003_0015.indd 8 24/02/18 15:44

Capitolo 1 Crescita cellulare e neoplasia 9
A oggi, quasi tutte le mutazioni ras nelle neoplasie del tratto GI si osservano nell’oncogene K-ras. La più alta frequenza di mutazioni viene riscontrata nei tumori esocrini del pancreas (>90%).27 Geni ras attivati attraverso mutazioni puntiformi sono stati identifi-cati approssimativamente nel 50% dei tumori maligni del colon, così come in un sottogruppo di tumori serrati (vedi Fig. 1-4).28
La maggior parte delle mutazioni di ras causa cambiamenti biochimici che lo mantengono nello stato attivo legato a GTP, riducendo l’attività della guanosintrifosfatasi (GTPasi) o desta-bilizzando la forma inattiva legata al GDP. Comunque, diversi mutanti ras conservano una significativa attività GTPasica; per-tanto, potrebbero essere coinvolti meccanismi aggiuntivi che con-vertono ras in una proteina oncogenica.29
Una conseguenza funzionale dell’attivazione di ras è la fosfo-rilazione di chinasi serina/treonina. Un importante bersaglio della cascata di segnalazione di ras è B-raf. Dei cancri del colon senza una mutazione K-ras identificabile, il 20% possiede una muta-zione attivante B-raf,30 confermando che l’attivazione di una via oncogena può essere raggiunta attraverso l’alterazione di una delle varie componenti sequenziali di una cascata di segnala-zione intra citoplasmatica.
Oncogeni nucleariAlcuni oncogeni codificano per proteine che si vanno a localiz-zare nel nucleo. In sostanza, questi prodotti oncogeni nucleari sono i mediatori finali dei pathway di trasduzione del segnale e agiscono come fattori trascrizionali che regolano l’espressione di geni che stimolano la proliferazione cellulare e sopprimono la normale differenziazione.
Il ruolo degli oncogeni nucleari è ben illustrato dalla famiglia myc. Il prodotto proteico c-Myc è coinvolto in funzioni cellulari critiche come la proliferazione, la differenziazione, l’apoptosi, la trasformazione e l’attivazione trascrizionale di geni chiave.31 Frequentemente, c-Myc è sovraespresso o amplificato in diversi cancri del tratto GI. c-Myc risulta essere un target trascrizionale del complesso β-catenina/TCF-4 nel cancro colorettale (vedi Fig. 1-3), in cui la sua espressione è significativamente aumentata.32
Geni oncosoppressoriI prodotti dei geni oncosoppressori prevengono l’acquisizione del fenotipo tumorale in vitro e hanno simili proprietà funzionali in vivo. Mutazioni che interrompono la funzione biologica di questi geni sono associate alla maggior parte dei tumori GI. Mutazioni della linea germinale di questa classe di geni sono alla base della maggior parte delle sindromi tumorali ereditarie dell’intestino, in cui uno specifico gene risulta alterato e i cui prodotti genici sono stati identificati e caratterizzati (Tab. 1-1).
Il riconoscimento dell’esistenza dei geni oncosoppressori è derivato dalle analisi genetiche di famiglie tendenti a sviluppare tumori. Nel tratto GI le sindromi ereditarie associate a cancro colorettale, gastrico e pancreatico sono quelle meglio descritte; queste sindromi sono discusse in altri capitoli del libro. Un certo numero di caratteristiche è comune in tutte le sindromi tumorali e presenta un’ereditarietà di tipo mendeliano. In particolare, un incremento marcato del rischio di sviluppare un particolare tumore è in questi casi presente in assenza di altri fattori ambientali pre-disponenti. In aggiunta, molteplici tumori primitivi si sviluppano spesso all’interno dello specifico tessuto e si manifestano a un’età più giovanile rispetto alla popolazione generale. Gli individui affetti sono inoltre a rischio di tumori al di fuori del tratto GI.
Queste osservazioni condussero Knudson a ipotizzare che i tumori, nelle sindromi familiari, potessero derivare da mutazioni indipendenti dei due alleli di uno specifico oncosoppressore (Fig. 1-5). Nello specifico, egli propose che la prima mutazione fosse presente in una copia del gene ereditato dalla linea germinale e, dunque, in tutte le cellule dei membri affetti della famiglia,33 men-tre una mutazione somatica nel rimanente allele normale del gene
TABELLA 1-1 Mutazioni associate alle sindromi tumorali ereditarie gastrointestinali
Disturbi Geni mutati
FAP, AFAP APC
Sindrome di Lynch (HNPCC) MSH2, MLH1, MSH6, PMS2, EpCAM
Poliposi MUTYH MUTYH
Sindrome di Peutz-Jeghers LKB1/STK11
Malattia di Cowden PTEN
Poliposi giovanile SMAD4, BMPR1A
Cancro gastrico diffuso ereditario
CDH1
Adenocarcinoma pancreatico ereditario
ATM, BRCA1, BRCA2, PALB2, PALLD, CDKN2A, PRSS1, SPINK1, PRSS2, CTRC, CFTR
MEN1 Menin
AFAP, FAP attenuata; APC, poliposi adenomatosa del colon; FAP, poliposi adenomatosa familiare; HNPCC, carcinoma colorettale ereditario non poliposico; MEN1, neoplasie endocrine multiple, tipo 1; MUTYH, omologo di mutY.
FIGURA 1-4. Teoria “dei due colpi” di Knudson. Nelle sindromi tumorali ereditarie, un cromosoma ha un locus per un gene oncosoppressore inattivo a causa di una mutazione della linea germinale. Il corrispon-dente gene oncosoppressore sul rimanente cromosoma accoppiato è conseguentemente inattivato da una mutazione somatica, portando alla formazione del tumore. Al contrario, nel cancro sporadico i due alleli del gene oncosoppressore vengono inattivati attraverso due mutazioni somatiche indipendenti, un evento improbabile all’interno di una sin-gola cellula.
Mutazionegerminale
Mutazionesomaticaacquisita
Mutazionesomaticaacquisita
Tumore
Secondamutazionesomatica
TumoreSindrometumoraleereditaria
Cancrosporadico
Tempo
X
X X
, Gene oncosoppressore
oncosoppressore si verificherebbe in una qualsiasi cellula e condur-rebbe allo sviluppo della neoplasia. Lo stesso gene potrebbe svolgere un ruolo nello sviluppo dello stesso tipo di tumore nella popolazione generale (cancro sporadico), ma sarebbero necessarie due mutazioni somatiche indipendenti di ciascuno dei due alleli. Tuttavia, questa combinazione di eventi dovrebbe essere poco comune e ciò spie-gherebbe la minore frequenza e la più avanzata età alla diagnosi di tumori similari nella popolazione generale. Comings fu il primo a suggerire che il gene rilevante in una sindrome cancerosa familiare potesse codificare per un prodotto genico di un oncosoppressore.34
001_SLEISENGER_01_0003_0015.indd 9 24/02/18 15:44

10 Sezione I Biologia del canale gastrointestinale
Anche se la teoria “dei due colpi” è stata generalmente osservata per le sindromi cancerose mendeliane, si tratta di eccezioni. Alcuni oncosoppressori possono determinare l’incremento del rischio onco-logico, in quanto un solo allele è mutato. Questi geni potrebbero essere così critici che la riduzione nell’espressione genica da parte di un allele mutato è sufficiente a guidare la tumorigenesi. In più, un allele mutante può operare in maniera dominante negativa, bloc-cando l’effetto di proteine codificate dall’allele normale.
Inattivazione dei geni oncosoppressoriAlcuni geni oncosoppressori furono inizialmente clonati attraverso il riconoscimento di porzioni di gene delete in campioni di tessuto tumorale provenienti da pazienti consanguinei con predisposizione allo sviluppo di neoplasie, mediante lo screening di DNA alla ricerca di marker dispersi nel genoma. Queste delezioni inattivano l’allele selvaggio e permettono di identificare la localizzazione sul cromo-soma del gene che causa la malattia, presente sull’altro allele. Più recentemente, la conoscenza delle variazioni genetiche osservate nei tumori è aumentata notevolmente mediante le tecnologie di nuova generazione di sequenziamento del DNA. Analizzando i cambiamenti genetici nei tumori e confrontandoli con la mucosa normale, siamo a conoscenza dei vari tipi di cambiamento genetico che avvengono nelle cellule tumorali. Le singole varianti nucleo-tidiche (Single Nucleotides Variants, SNVs) si riferiscono a cam-biamenti in una singola coppia di basi nel codice genetico. Mentre molte di queste mutazioni sono silenti, altre possono produrre significativi cambiamenti nell’espressione e nella funzione genica. Le mutazioni missense determinano un cambiamento nell’amino-acido codificato dal codone. Le mutazioni nonsense si riferiscono all’introduzione anticipata di un codone di stop. Le SNVs nei siti slice-acceptor o nei siti donatori possono indurre perdita di esoni o alterata espressione di sequenze introniche. Le SNVs nella sequenza promoter o nelle regioni regolatorie non trascritte di un gene pos-sono cambiarne drammaticamente l’espressione genica. Altri tipi di mutazioni genetiche comprendono le inserzioni e le delezioni. Mutazioni che prevedono inserzioni o delezioni di piccole sequenze di DNA possono determinare mutazioni frameshift all’interno di un gene. Tuttavia, sono state descritte anche inserzioni o delezioni di grosse porzioni di DNA. Tutte queste mutazioni possono risul-tare nell’inattivazione di un dato gene e rappresentano importanti meccanismi di inattivazione di uno dei due alleli del gene onco-soppressore. Un altro meccanismo di inattivazione di un gene oncosoppressore prevede l’inattivazione della porzione promoter del gene. Il silenziamento trascrizionale può derivare dalla meti-lazione delle sequenze CpG nei promoter del gene; ciò è stato dimostrato nei geni che codificano per p16INK4A ed E-caderina.35 Un’esagerata metilazione delle sequenze CpG è stata identificata come una caratteristica peculiare nelle vie che portano al cancro del colon (vedi Fig. 1-4).
I geni oncosoppressori non funzionano alla stessa maniera in ogni tipo di tessuto. Di conseguenza, l’inattivazione di un parti-colare gene oncosoppressore è tumorigenica solo in determinati tessuti. Per esempio, i geni oncosoppressori RB1 e VHL svolgono un ruolo determinante rispettivamente nel retinoblastoma e nel cancro del rene a cellule chiare, ma sono raramente mutate nelle neoplasie del tratto GI. Tre geni oncosoppressori sembrano avere un ruolo importante nella patogenesi delle neoplasie del tratto GI: APC, TP53 e SMAD4.
Gene Adenomatous Polyposis Coli (APC)Studi genetici di “linkage” hanno rivelato la presenza di marker sul cromosoma 5q21 che erano strettamente correlati allo sviluppo di polipi in membri della stessa famiglia con poliposi adenomatosa familiare (Familial Adenomatous Polyposis, FAP) e sindrome di Gardner.36 Successivi studi condussero all’identificazione del gene responsabile della FAP, il gene Adenomatous Polyposis Coli (APC).37-39 Per un’analisi dettagliata delle sindromi collegate alla poliposi adenomatosa, coinvolgenti il gene APC, si rimanda al Capitolo 126.
Mutazioni somatiche del gene APC sono state identificate anche nella maggior parte delle forme sporadiche di poliposi e di cancro del colon.40,41 Mutazioni del gene APC sono caratteristiche nelle forme precoci di adenoma, indicando un ruolo determinante di APC come gatekeeper nella progressione multistep da epitelio nor-male a cancro del colon (vedi Fig. 1-4).
Il gene APC comprende 15 esoni e codifica per una proteina di 2843 aminoacidi, del peso approssimativo di 310 kD. La maggior parte delle mutazioni del gene APC, germinali o somatiche, deter-mina la comparsa di codoni di stop prematuri e quindi la produzione di una proteina APC tronca. Le mutazioni che avvengono nella porzione aminoterminale della proteina APC sono associate a una rara variante di FAP, denominata poliposi adenomatosa familiare attenuata (AFAP).42 Le mutazioni di APC conducono a cambiamenti funzionali nelle interazioni proteina-proteina. Come discusso pre-cedentemente, APC è un regolatore negativo del pathway di segna-lazione Wnt (vedi Fig. 1-3). Le proteine APC mutate sono incapaci di interagire con la β-catenina e da ciò risulta un’attivazione incon-trollata del pathway Wnt e il conseguente fenotipo oncogenico.
Gene TP53Così chiamato perché produce una proteina del peso di 53 kD, p53 è una fosfoproteina nucleare che svolge un ruolo chiave nella regolazione del ciclo cellulare e nell’apoptosi.43 La proteina p53 fu identificata per la prima volta nei tumori quale prodotto di un gene mutato che risiedeva nel cromosoma 17p, una regione che manifesta una perdita di eterozigosi in molti tumori. Mutazioni puntiformi di TP53 sono identificate nel 50-70% dei casi di cancro del colon sporadico (vedi Fig. 1-4), ma solamente in un piccolo gruppo di adenomi del colon.44 Mutazioni puntiformi di TP53 si trovano in tutte le neoplasie maligne del tratto GI.43 È da sottoli-neare che l’aflatossina sembra indurre una mutazione nel codone 249 di TP53 in molti carcinomi epatocellulari.45 In aggiunta alle mutazioni puntiformi di TP53 nelle neoplasie maligne sporadiche, mutazioni germinali di TP53 sono state osservate nella sindrome di Li-Fraumeni, una patologia familiare autosomica dominante in cui le persone affette possono sviluppare carcinoma della mam-mella, sarcoma dei tessuti molli, osteosarcoma, leucemia, tumori del cervello e carcinomi surrenalici.46
La proteina p53 è indotta in condizioni di stress cellulare, come quello prodotto da radiazioni ionizzanti, mancanza di fattori di crescita o terapia citotossica (vedi Fig. 1-2). A seguito del danno genotossico, p53 determina arresto della proliferazione cellulare alla fase G1 per facilitare la riparazione del DNA o fenomeni di sene-scenza, oppure per attivare meccanismi apoptotici. Quindi, p53 media alcune di queste risposte mediante l’induzione dell’inibitore del ciclo cellulare p21CIP1/WAF1 o di geni proapoptotici, come PUMA, e c-Myc sembra svolgere un ruolo nel determinare il destino della cellula.47
Gene SMAD4SMAD4 è un gene oncosoppressore localizzato sul cromosoma 18q. Questo gene è deleto o mutato nella maggior parte degli adenocar-cinomi del pancreas e in un sottogruppo di neoplasie maligne del colon. Esso codifica per la proteina Smad4, un mediatore intracellulare essenziale degli effetti inibitori sulla crescita cellulare di TGF-β. La proteina Smad4 ha due importanti domini, mad homology domain 1 e 2 (MH1 e MH2), che sono essenziali per il legame al DNA e per promuovere la formazione di oligomeri con altre proteine Smad.48 La proteina Smad4 mutata blocca l’inibizione della proliferazione indotta da TGF-β. Mutazioni della linea germinale di SMAD4 esi-tano nella sindrome da poliposi giovanile (vedi Cap. 126).
Geni deputati alla riparazione del DNAI meccanismi cellulari si sono evoluti per mantenere l’integrità del DNA. Errori possono essere introdotti nel genoma attraverso mol-teplici meccanismi fisiologici e patologici. Questi errori compren-dono il mismatching spontaneo dei nucleotidi durante la normale
001_SLEISENGER_01_0003_0015.indd 10 24/02/18 15:44

Capitolo 1 Crescita cellulare e neoplasia 11
replicazione del DNA, il danno ossidativo dei nucleotidi e la rot-tura completa del doppio filamento. Esistono numerosi sistemi distinti per riparare questi tipi di alterazioni del DNA che pos-sono originarsi a seguito di numerosi tipi di danno, tra cui carci-nogeni, radiazioni ionizzanti e radicali liberi dell’ossigeno. Un tipo di errore che si sviluppa durante la replicazione può verificarsi in filamenti di DNA microsatellite, che comprende regioni di ripeti-zioni mononucleotidiche (per es., poly-A) o dinucleotidiche (per es., poly-CA).49 Il sistema DNA mismatch repair corregge questi errori. Gli enzimi si legano al DNA mismatched I, tagliano il filamento di DNA con il nucleotide interessato, svolgono il frammento, riem-piono il gap con il nucleotide corretto e, infine, risigillano il fila-mento. La famiglia di geni deputati alla riparazione del mismatch comprende MSH2, MSH3, MSH5, MSH6, MLH1, PMS1 e PMS2.
MLH1 e MSH2 sono i due geni che riparano il DNA mismatch più frequentemente mutati a livello germinale nella sindrome di Lynch, anche conosciuta come cancro colorettale ereditario non poliposico (HNPCC).50,51 Le mutazioni possono condurre ad alterazioni funzio-nali che producono slippage del filamento durante la replicazione. Le cellule interessate sono chiamate Replication ERror (RER)-positive, in contrapposizione al fenotipo RER-negativo.52,53 Poiché le sequenze di DNA microsatellite sono principalmente interessate da questo tipo di instabilità genetica, le cellule tumorali sono conseguente-mente dette portatrici di instabilità del microsatellite (MicroSatellite Instability, MSI). Dal punto di vista meccanicistico, la mancanza di sistemi di riparazione del DNA non causa direttamente il cancro. Piuttosto, il difetto di tali meccanismi di riparazione crea un sub-strato che produce l’accumulo di mutazioni in una grande varietà di altri geni che contengono sequenze di DNA microsatellite, come per esempio il recettore per il TGF-β di tipo 2, il recettore per l’IGF tipo 2, BAX e E2F-4. L’instabilità dei microsatelliti rappresenta un nuovo meccanismo per l’accumulo di mutazioni all’interno di un tumore (vedi Fig. 1-4). Questa alterazione è caratteristica di tutti i tumori collegati alla sindrome di Lynch e si osserva approssima-tivamente nel 15% di tutti i cancri del colon sporadici. Evidenze sempre più numerose dimostrano che questi tumori possiedono le caratteristiche dell’adenoma serrato e presentano un’ipermeti-lazione del promotore del gene MLH1 (vedi Fig. 1-4).
Errori possono essere anche introdotti quando singoli nucleotidi sono danneggiati da sostanze chimiche ed esistono sistemi definiti base excision repair che correggono questo tipo di errori. Residui di 8-oxoguanina possono derivare da stress ossidativo del DNA e tali basi alterate si appaiano in maniera impropria con basi di adenina che, infine, conducono a mutazioni somatiche G:C→T:A. MUTYH è una DNA-glicosilasi che è coinvolta nella riparazione di questi nucleotidi di guanina ossidati. È stata identificata una sindrome poliposica adenomatosa autosomica recessiva causata da mutazioni germinali del gene MUTYH.54,55 Da notare che le mutazioni G:C→T:A del gene APC sono quasi universalmente identificate nei polipi di pazienti con mutazioni geminali MUTYH, indicando l’esistenza di importanti similitudini nella patogenesi molecolare della poliposi nelle sindromi MUTYH e FAP.
Vie di segnalazione degli oncogeniSingoli oncogeni o geni oncosoppressori non necessariamente indu-cono direttamente la trasformazione cellulare, ma più semplicemente operano come componenti di un grande network di segnalazione tumorale. Alcuni pathway particolarmente rilevanti per la genesi dei tumori GI comprendono le vie di segnalazione Wnt e Ras. Queste sono vie che regolano la normale omeostasi tissutale, ma diventano oncogeniche quando i segnali sono trasdotti in maniera aberrante o amplificata. Gli aspetti chiave delle vie di segnalazione Wnt sono illustrate nella Figura 1-3. La β-catenina si porta dalla membrana plasmatica interna al citoplasma e qui forma un complesso macro-molecolare con la proteina APC Axin e la glicogeno sintasi china-si-3β (GSK-3β). La fosforilazione della β-catenina da parte della GSK-3β stimola la sua degradazione. In presenza di un segnale Wnt attivo, la β-catenina è stabilizzata, entra nel nucleo e interagisce con
il fattore di trascrizione Tcf-4 determinando la sovraregolazione di un certo numero di geni target, inclusi c-Myc, cyclin D1 e VEGF. Come discusso precedentemente, la via di segnalazione collegata a Wnt è essenziale per la regolazione della proliferazione dell’epi-telio intestinale normale ed è alterata in quasi tutti i tumori maligni del colon-retto. Ciò può derivare da mutazioni dei geni APC, Axin o b-catenina, sebbene le alterazioni del gene oncosoppressore APC siano le più comuni. L’alterazione di uno solo di questi componenti è sufficiente per attivare l’intero pathway. Quindi, è importante contestualizzare queste singole alterazioni genetiche nel network di segnale in cui esse svolgono la loro funzione.
Siccome questi pathway sono tipicamente non lineari, ne deri-vano livelli aggiuntivi di complessità. Infatti, è frequente una sovrap-posizione fra queste vie, al punto che una distinzione può essere talvolta arbitraria. Per esempio, mutazioni dell’oncogene K-ras deter-minano l’attivazione di multiple e distinte vie, comprendenti Raf/ERK/MAPK, PI3K/Akt e NF-κB, ciascuna delle quali svolge un ruolo importante nella tumorigenesi (Fig. 1-6). Il crosstalk fra que-ste vie effettrici serve a modulare ulteriormente le risposte cellulari. Per esempio, Akt, un substrato PI3K, può fosforilare Raf e quindi regolare il signaling attraverso la via di segnale di MAPK.56 Infine, ciascuna di queste vie di segnalazione regola molteplici processi biologici correlati alla tumorogenesi,57 tra i quali la progressione del ciclo cellulare, l’apoptosi, la senescenza, l’angiogenesi e l’invasione.
Un’altra via che svolge un ruolo particolarmente importante nei tumori GI è quella della ciclo-ossigenasi-2 (COX-2). L’enzima COX-2 è un regolatore chiave della sintesi di prostaglandine ed è indotto nell’infiammazione e nelle neoplasie. Sebbene nessuna mutazione di COX-2 sia stata descritta, la sovraespressione di COX-2 negli adenomi e nelle neoplasie maligne del colon è asso-ciata alla progressione tumorale e all’angiogenesi, principalmente attraverso la sintesi di prostaglandina E2. L’inibizione della COX-2 da parte di numerosi farmaci (acido acetilsalicilico, FANS o inibitori selettivi della COX-2) è associata a un minor rischio di adenomi o cancro colorettale.58
RNA non codificantiRecentemente, è stato chiarito che una significativa porzione del genoma non codificante per proteine rimane attivo dal punto di vista trascrizionale. L’RNA, denominato non-coding RNA (ncR-NAs), consiste di un’estesa categoria di molecole di RNA attivo che
FIGURA 1-5. Diversità delle vie di segnalazione tramite K-ras. La variante oncogenica di K-ras può attivare multipli pathway di segnalazione. I meccanismi che determinano quale via può venire preferenzial-mente attivata in una data cellula non sono ancora del tutto chiari. Il crosstalk fra queste vie di segnalazione incrementa la complessità della rete di signaling. Queste vie di segnalazione effettrici possono influenzare vari processi biologici della cellula, tra cui la proliferazione, l’apoptosi, la differenziazione e la motilità.
K-ras
RALGDS
Ral
Raf
MEK
ERK/MAPK
Akt
PI3K JNK NF-kB
AP-1
001_SLEISENGER_01_0003_0015.indd 11 24/02/18 15:44

12 Sezione I Biologia del canale gastrointestinale
contengono long non-coding RNA (lncRNA) e micro-RNA (miRNA), che sono frequentemente alterati nelle neoplasie.59 Inizialmente modificati in small interfering RNA (siRNA) da parte della proteina Dicer in sequenze di 20-25 nucleotidi, i microRNA svolgono un ruolo importante nel silenziamento dei trascritti genici.60 Gli siRNA si legano alle sequenze complementari di mRNA e questo legame facilita l’attività dei sistemi deputati al taglio e alla degradazione dell’mRNA. Il lncRNA può svolgere diverse funzioni, quali il silenziamento genico, lo splicing e l’estensione dei telomeri.
EpigeneticaL’epigenetica si riferisce a cambiamenti nel genoma che determi-nano modificazioni del fenotipo senza cambiamenti effettivi nella sequenza del DNA. Spesso questi cambiamenti possono risul-tare da alterazioni strutturali della sequenza del DNA. Uno dei meccanismi più importanti è l’ipermetilazione della sequenza CpG nei promotori genici. I promotori di molti geni sono ricchi di sequenze CpG (isole CpG). La metilazione dei residui di citosina in questi siti può determinare il silenziamento del gene a valle.
Molte neoplasie maligne mostrano ipermetilazione del promo-tore e silenziamento di importanti geni oncosoppressori. In circa il 15-20% dei casi di cancro del colon-retto, questo processo diventa un fattore determinante della carcinogenesi. Caratterizzati come CpG Island Methylator Phenotype (CIMP)-positivi, questi tumori hanno eccessivi livelli di ipermetilazione del promotore del gene oncosop-pressore. Va sottolineato il fatto che il gene MLH1 è frequentemente ipermetilato, portando allo sviluppo di tumori maligni sporadici con instabilità dei microsatelliti. I meccanismi alla base di questi processi di ipermetilazione del promotore rimangono sconosciuti, ma recenti studi dimostrano un legame tra il metabolismo tumo-rale e lo stato di metilazione. Mutazioni del gene IDH1 possono indurre un fenotipo CIMP-positivo nei glioblastomi.61
METABOLISMO DEL TUMOREGli stimoli metabolici e la disponibilità di nutrienti rivestono un ruolo fondamentale nell’omeostasi e nella crescita cellulare. Come descritto in precedenza, una carenza di metaboliti o una disfun-zione mitocondriale può indurre arresto della crescita o apoptosi. Tuttavia, le cellule neoplastiche mostrano assetti metabolici ano-mali che ne agevolano la crescita e le necessità anaboliche. Studi condotti nel 1924 dal premio Nobel Otto Heinrich Warburg rive-larono che le cellule tumorali manifestano un’importante acce-lerazione nei processi di glicolisi aerobia e una riduzione della respirazione mitocondriale. Quest’ipotesi, definita come l’ipotesi di Warburg, è stata dimostrata ed è un elemento caratteristico di molte neoplasie maligne.62 Molti dei geni coinvolti nelle neo-plasie del tratto GI (p53, K-Ras, PI3K, mTOR, HIF, Myc) svolgono una funzione regolatoria in molti pathway metabolici. Inoltre, mutazioni germinali di geni regolatori del metabolismo (per es., quelli per le subunità della succinato deidrogenasi [SDH]), che normalmente non sono oncogeni o geni oncosoppressori in senso stretto, sono stati associati a un alto rischio oncogenico (feocromo-citoma e paraganglioma).63,64 Il vantaggio selettivo che un incre-mento della glicolisi porta alla cellula tumorale può includere la maggiore resistenza in ambiente ipossico e l’allontanamento di sottoprodotti di altre vie metaboliche. Queste vie metaboliche alterate rappresentano nuovi target terapeutici promettenti.
INFLUENZE AMBIENTALI E MICROAMBIENTALIDi base, il cancro è una malattia genetica. I fattori ambientali svolgono un ruolo cruciale nell’oncogenesi, ma, in definitiva, portano all’espressione di geni alterati o all’iperespressione di
geni normali i cui prodotti conferiscono il fenotipo maligno. La mutazione genetica rappresenta il comune denominatore dell’ef-fetto degli agenti che contribuiscono allo sviluppo della neoplasia.
Carcinogenesi chimicaL’attivazione metabolica dell’ospite è un fattore chiave nel potenziale oncogeno di molti composti chimici. Il composto iniziale, il procarcinogeno, è convertito dagli enzimi dell’ospite in derivati elettrofili che alterano la struttura chimica del DNA. Le mutazioni originano da errori che accadono durante la replicazione del DNA come risultato di coppie di basi azotate distorte. Fattori che influen-zano il potenziale tumorale di ogni carcinogeno chimico inclu-dono l’equilibrio esistente fra l’attivazione del procarcinogeno e la disattivazione o degradazione del carcinogeno.65 La disattiva-zione si verifica tipicamente attraverso reazioni di coniugazione, solitamente nel fegato.
Questi principi sono esemplificati dai carcinomi del colon che si sviluppano in ambito sperimentale in topi nutriti con cicasina, un composto glicosilato contenuto nelle noci di cicas. I residui di glucosio vengono scissi nel fegato del ratto da una β-glucosi-dasi per formare metilazossimetanolo, che è successivamente deformilato da enzimi presenti nel fegato e nel colon a formare metildiazionio, che costituisce un potente carcinogeno. Questi stessi metaboliti sono prodotti nel fegato tramite modificazione enzimatica del composto dimetilidrazina, dando origine a neo-plasie maligne del colon nel ratto.
Nell’uomo, l’uso assiduo di tabacco è fortemente associato allo sviluppo di diversi tipi di carcinomi del tratto GI, inclusi il carcinoma colico e pancreatico. Nei fumatori attivi e con lunga storia di esposizione al tabacco, il rischio di cancro del pancreas può raddoppiare. Molteplici agenti cancerogeni, tra cui l’arse-nico, il benzene e l’ossido di etilene, sono stati identificati nelle sigarette, ma gli agenti chimici collegati in maniera specifica allo sviluppo del cancro del pancreas o del colon non sono ancora stati definiti.
Fattori dieteticiLa mutagenesi chimica può essere particolarmente importante nello sviluppo di carcinomi nell’ambito del tratto GI e negli organi correlati. Le mucose, da cui originano la maggior parte delle neo-plasie primitive GI, sono esposte a una complessa miscela di costituenti dietetici che rappresentano potenziali carcinogeni o procarcinogeni. La capacità dei fattori dietetici di agire come mutageni è stata direttamente dimostrata nel 1995. La frequenza di contaminazione dei cibi con aflatossine, metaboliti fungini, risulta correlata all’incidenza di epatocarcinoma in diverse aree del mondo.66 Gli studi che hanno dimostrato che le aflatossine causano la mutazione del gene TP53 nell’epatocarcinoma hanno fornito un collegamento stringente fra geni e ambiente.66
I nitrati presenti in molti cibi sembrano essere ulteriori elementi dietetici che possono agire come procarcinogeni nel tratto GI. I nitrati derivati dagli alimenti possono essere convertiti dall’at-tività batterica, in un ambiente gastrico ipocloridrico, in nitriti e, successivamente, in nitrosamine ad attività mutagena.67 Questi eventi possono essere alla base della dimostrata correlazione fra l’assunzione di cibi ricchi di nitrati e l’incidenza del cancro gastrico in popolazioni differenti.
Altri fattori dietetici possono modulare la potenza biologica dei procarcinogeni alimentari. Variazioni nelle quantità assolute o relative dei grassi assunti con la dieta possono portare a un’al-terazione dei componenti della flora microbica del colon e delle loro caratteristiche metaboliche, risultando in una modulazione della produzione di enzimi che convertono i costituenti della dieta in composti potenzialmente mutageni. Cambiamenti nella quantità di fibre contenute nella dieta possono alterare il tempo di transito del contenuto intestinale, cambiando così il tempo
001_SLEISENGER_01_0003_0015.indd 12 24/02/18 15:44

Capitolo 1 Crescita cellulare e neoplasia 13
di esposizione della mucosa ai potenziali mutageni contenuti nel lume. Il contenuto luminale di sali biliari può essere un fat-tore addizionale in grado di modulare l’effetto biologico dei procarcinogeni. I sali biliari non coniugati possono promuovere la carcinogenesi attraverso un danno mucosale e un’aumentata proliferazione epiteliale.
Questi meccanismi possono spiegare la ben documentata cor-relazione tra l’assunzione di diversi componenti della dieta e l’in-cidenza di cancro del colon-retto in determinate popolazioni. Le popolazioni in cui l’apporto di fibre è elevato, con conseguente riduzione del tempo di transito colico, generalmente mostrano una minore incidenza di cancro del colon retto rispetto a quelle con ridotta assunzione di fibre e conseguente transito rallentato. L’incidenza di cancro del colon retto nei giapponesi immigrati negli Stati Uniti, che seguono una dieta occidentale, è molto più alta rispetto a quella dei giapponesi nativi che seguono la dieta tradizionale locale.68
MicrobiomaL’organismo umano alberga più di 100 mila miliardi di microbi. Le interazioni fra questi organismi e l’ospite è un argomento di grande interesse, in particolare per un ampio spettro di malattie autoimmuni, metaboliche e neoplastiche. Lo Human Microbiome Project sta cercando di sviluppare una mappa di questi organi-smi dispersi ovunque nel nostro corpo, con l’obiettivo di corre-lare specifiche specie batteriche con determinati quadri patolo-gici. Sebbene i risultati di questo filone di ricerca siano ancora preliminari, vi sono sempre più evidenze che la composizione del microbiota intestinale può influenzare il rischio di cancro.69 Popolazioni batteriche alterate hanno la possibilità di influenzare pathway metabolici e indici infiammatori nel tratto GI.
Anche i virus possono alterare i normali geni tramite l’in-tegrazione nel genoma dell’ospite in posizioni che alterano la normale sequenza genica (mutagenesi inserzionale) o attraverso l’introduzione di geni aberranti presenti nel patrimonio genetico virale. I virus che sembrano rivestire un ruolo nell’oncogenesi del tratto GI sono il papillomavirus nei carcinomi squamocellulari dell’esofago e dell’ano, il virus di Epstein-Barr nei tumori lin-foepiteliali gastrici e il virus dell’epatite B nell’epatocarcinoma.
Cancro e infiammazioneUn certo numero di quadri infiammatori cronici aumenta il rischio sito-specifico di cancro: colite ulcerosa (vedi Cap. 116), gastrite cronica (vedi Cap. 52), pancreatite cronica (vedi Cap. 59), esofago di Barrett (vedi Cap. 45), epatiti virali croniche (vedi Capp. 79 e 80). In aggiunta allo stimolo proliferativo, l’influenza dell’infiam-mazione nello sviluppo della neoplasia è complessa e sfaccettata. Le cellule del sistema immunitario possono indurre il rimodel-lamento della rete vascolare e promuovere l’angiogenesi (vedi oltre). L’infiammazione può anche indurre alterazioni epigeneti-che che favoriscono l’inibizione di geni oncosoppressori, tramite un danneggiamento del DNA legato alla produzione di radicali liberi mediata dalle citochine. Inoltre, le citochine prodotte dalle cellule infiammatorie possono portare all’attivazione, nelle cellule tumorali, del fattore nucleare (NF)-κB, che può portare all’inibi-zione dell’apoptosi e stimolare la proliferazione.70 Sebbene l’in-fiammazione crei un ambiente pro-oncogenico, va tenuto conto che il sistema immunitario svolge un ruolo importante anche nella soppressione delle neoplasie tramite la sorveglianza tumo-rale. Le terapie immunosoppressive sono associate a un aumento del rischio di cancro. Il mantenimento di questo preciso bilan-cio immunoregolatorio è fondamentale nella prevenzione dello sviluppo di un ambiente pro-oncogenico.
ASPETTI BIOLOGICI DELLE METASTASI TUMORALIL’attecchimento di metastasi a distanza richiede processi multi-pli, molti dei quali riguardano l’alterazione delle interazioni tra la cellula tumorale e le cellule dell’ospite. Per metastatizzare, una cellula o un gruppo di cellule deve staccarsi dal tumore primitivo, accedere agli spazi vascolari o linfatici, aderire alla superficie endoteliale di una zona distante, penetrare la parete vasale per invadere il sito secondario e, infine, proliferare come un secondo focus neoplastico. L’angiogenesi è necessaria per la proliferazione del tumore primario e delle metastasi. Le cellule neoplastiche devono anche sopraffare i meccanismi immunitari di killing cellulare. Di conseguenza, poche cellule tumorali circo-lanti (<0,01%) sono in grado di dare il via a nuovi foci metastatici. È stata proposta, riguardo alle metastasi, una visione “survival of the fittest”, in cui la competizione selettiva favorisce metastasi appartenenti a diverse sottopopolazioni di cellule originarie dal tumore primitivo.71 Dopo la formazione di un nuovo focus meta-statico si realizza nuovamente un’espansione clonale.
Transizione epitelio-mesenchimaLa modulazione delle interazioni delle cellule tumorali con le cellule adiacenti e con la matrice extracellulare è un passaggio essenziale, in quanto le cellule tumorali epiteliali attraversano la membrana basale e infine metastatizzano a distanza. Un processo simile si manifesta durante l’embriogenesi, quando le cellule epiteliali pola-rizzate non riconoscono più i confini imposti dalle cellule epiteliali vicine o dalla loro membrana basale e assumono le caratteristiche di cellule mesenchimali migranti. Questo fenomeno, denominato transizione epitelio-mesenchima (Epithelial-Mesenchymal Transition, EMT), ha fornito degli spunti di ragionamento per la compren-sione della progressione tumorale (Fig. 1-7). L’E-caderina è un componente cruciale delle giunzioni aderenti che mantengono le
FIGURA 1-6. Una transizione epitelio-mesenchima (EMT) fornisce un modello per la progressione e l’invasione tumorale. Le cellule epiteliali mantengono la loro polarità e i contatti con le cellule adiacenti tramite molte proteine giunzionali, compresa la E-caderina. La perdita o la sottoregolazione della E-caderina è una caratteristica fondamentale della EMT, in cui le cellule epiteliali acquisiscono un fenotipo mesen-chimale di tipo migrante. Nella progressione tumorale, la EMT può manifestarsi a diversi livelli, tra cui la transizione tra il carcinoma in situ e cancro invasivo e l’invasione dei vasi ematici e linfatici da parte delle cellule tumorali.
Membranabasale
Stroma
Displasia di altogrado/carcinoma in situ
Perditadi E-caderina
EMT
EMT
MetastasiVasi sanguigni
001_SLEISENGER_01_0003_0015.indd 13 24/02/18 15:44

14 Sezione I Biologia del canale gastrointestinale
interazioni tra le cellule epiteliali, e la perdita di E-caderina è una delle caratteristiche fondamentali del fenotipo EMT.72 Mutazioni della E-caderina sono comuni in molte neoplasie GI, in particolare nel cancro gastrico. Mutazioni germinali della E-caderina sono correlate al cancro gastrico diffuso ereditario.
La membrana basale epiteliale è formata da una densa matrice di collagene, glicoproteine e proteoglicani che, normalmente, non permette la penetrazione passiva delle cellule. La trasmigrazione delle cellule tumorali attraverso la membrana basale verosimilmente implica la generazione di attività proteolitiche chiave. In alternativa, le cellule tumorali possono produrre fattori capaci di attivare dei proenzimi presenti nella matrice extracellulare, come per esempio l’urochinasi o l’attivatore del plasminogeno. Avendo guadagnato l’ac-cesso al compartimento stromale intestinale, le cellule tumorali pos-sono quindi penetrare nei vasi linfatici e sanguigni e metastatizzare.
Angiogenesi e linfoangiogenesiL’angiogenesi è fondamentale per sostenere la crescita continua del tumore primitivo. Se non si formano nuovi vasi via via che il tumore primitivo si espande, le cellule più distanti dai vasi dispo-nibili sono private di un’adeguata fonte di nutrienti, con conse-guente necrosi della parte centrale del tumore. La neovascolariz-zazione è anche un importante fattore permissivo nel facilitare la disseminazione metastatica dei tumori.73 Diversi fattori di crescita proteici sono stati descritti come potenti stimoli angiogenetici, tra cui il fattore di crescita dell’endotelio vascolare (Vascular Endothelial Growth Factor, VEGF)-A, il fattore di crescita basico dei fibroblasti (basic Fibroblast Growth Factor, bFGF) e il TGF-β. Il VEGF-A è forse il fattore più decisivo, sovraregolato in molti tipi di tumore, incluso il cancro colorettale. Multipli pathway genetici implicati nell’oncogenesi GI modulano l’espressione di VEGF-A, tra cui Wnt e le mutazioni di ras.74 Strategie volte a ini-bire VEGF-A fanno parte delle linee-guida terapeutiche per il cancro del colon retto metastatico (vedi Cap. 127).
L’angiogenesi si verifica attraverso una serie ordinata di eventi. Le cellule endoteliali del vaso principale sono stimolate a degradare la membrana basale endoteliale, migrare nello stroma perivascolare e dare il via alla formazione di una sorta di germoglio vascolare che si accresce e si trasforma in una struttura tubulare, dando origine a una nuova rete capillare. I modelli in vitro che riproducono le fasi precoci dell’angiogenesi indicano che questo processo si attua tramite un bilancio fra proteasi e inibitori delle proteasi, con modalità simili a quelle che si osservano nei processi di invasione tumorale. Esistono, infatti, dei parallelismi funzio-nali tra invasione tumorale e angiogenesi, quali la proteolisi della membrana basale, il movimento, l’invasione e la crescita cellulare.
In aggiunta all’angiogenesi, anche la linfoangiogenesi ha un ruolo critico nei processi di metastatizzazione tumorale. Sono state acquisite alcune importanti nozioni sulle basi molecolari della linfoangiogenesi tumorale. VEGF-C, o VEGF-D, si lega al recettore 3 del VEGF sulle cellule linfatiche endoteliali per stimo-lare la formazione di nuovi vasi linfatici.75 Questo conduce alla produzione di nuovi vasi linfatici nell’ambito nella massa tumorale e, conseguentemente, alla disseminazione delle cellule cancerose ai linfonodi regionali.76 Strategie volte a inibire la linfoangiogenesi tumorale sono attualmente oggetto di studio.
MEDICINA MOLECOLARE: APPROCCI ATTUALI E FUTURI ALL’ONCOLOGIA GASTROINTESTINALE
Diagnostica molecolareI progressi compiuti nell’identificazione dei geni correlati al tumore, insieme alle potenzialità intrinseche delle tecniche di biologia molecolare di analizzare quantità di DNA o di proteine
incredibilmente piccole, stanno portando all’individuazione di marker diagnostici più efficaci rispetto al passato. L’applicazione più immediata è la valutazione del rischio di cancro in mem-bri di famiglie con predisposizione alle malattie tumorali. Sono state sviluppate procedure per identificare mutazioni germinali in pazienti con varie sindromi oncologiche GI ereditarie, com-prese la FAP, la sindrome di Lynch e il cancro gastrico diffuso ereditario (HDGC) (vedi Tab. 1-1). I test genetici sono uno stru-mento molto utile per identificare il rischio oncologico nei sin-goli membri di una famiglia. L’applicazione dei test genetici deve tenere conto sia della sensibilità e della specificità dell’analisi sia delle implicazioni in termini medico-legali. Per queste ragioni, il counseling genetico è un passo essenziale del processo di valu-tazione genetica.
Una migliore capacità di rilevazione dei tumori GI spora-dici e delle lesioni precancerose associate è da molti anni l’obiet-tivo della ricerca oncologica GI. Un piccolo numero di cellule di sfaldamento ottenute dalle feci o dall’aspirazione di fluido dalle lesioni cistiche possono essere analizzate per ricercare la presenza di mutazioni o di alterazioni epigenetiche in geni specifici cor-relati ai tumori (B-raf, K-ras, APC, TP53 ecc.). Test MSI possono essere eseguiti su preparati di tumori colici, come screening volto a identificare gli individui in cui i tumori colorettali possono essersi sviluppati nell’ambito della sindrome di Lynch o di altre sindromi tumorali del colon-retto.77 La perdita della positività immunoistochimica per MSH2, MLH1, PMS2 o MSH6 può fornire importanti informazioni. Vari studi hanno dimostrato che lo sta-tus MSI di un tumore del colon è un fattore predittivo di risposta alla chemioterapia con 5-fluorouracile.78,79 Inoltre, terapie dirette contro specifiche vie di segnalazione permetteranno, parallela-mente a una migliore comprensione molecolare dei tumori GI, l’attuazione di terapie personalizzate, che attualmente costitui-sce l’obiettivo primario in ambito oncologico. Anticorpi in grado di legare i recettori di EGF e di bloccarne la via di segnalazione hanno apportato un beneficio significativo nella cura del cancro del colon-retto, ma solo in pazienti che non presentavano mutazioni attivanti di K-ras. In quest’ottica, la ricerca di mutazioni di K-ras nei tumori del colon-retto viene eseguita di routine prima di uti-lizzare tali terapie mirate. Inoltre, l’inibitore della tirosinchinasi dell’oncogene c-KIT costituisce oggi un trattamento di routine per i tumori stromali GI (vedi Cap. 32).80 Le tecnologie molecolari possono anche trovare applicazione nello staging delle neoplasie. Per esempio, la ricerca di cellule tumorali circolanti prima dell’in-dividuazione delle metastasi potrebbe portare a notevoli benefici sia prognostici sia terapeutici.81 La progressiva disponibilità di test per marker genetici permetterà un sensibile miglioramento del monitoraggio della ripresa di malattia dopo l’eradicazione chirurgica di una neoplasia.
Studi di genome-wide associationSebbene l’11% degli individui con cancro del colon-retto abbia due parenti stretti affetti dalla medesima malattia, solo una piccola parte di casi rientra nell’ambito di sindromi oncologiche genetiche a trasmissione mendeliana già definite.82 Inoltre, studi sul cancro colorettale in gemelli omozigoti hanno dimostrato che il rischio non supera il 35%. L’identificazione di altre varianti genetiche che conferiscono un rischio aumentato di cancro del colon-retto rimane un’importante priorità in ambito diagnostico. Con lo sviluppo di tecnologie di genotipizzazione e di sequenziamento genico sono state scoperte molte altre varianti. Due ipotesi hanno indi-rizzato le ricerche di queste varianti. La prima, definita “malattia comune-variante comune”, è basata sull’idea che il rischio eredi-tabile per malattie come il cancro del colon-retto sia basato sulla somma di piccoli effetti di varianti genetiche frequenti (alleli a frequenza minore >5%) nella popolazione generale. Finora, molti loci genici sono stati identificati. La quota di rischio relativo di ogni variante associata non ha portato ad alcuna ulteriore infor-mazione con valore predittivo aggiuntivo rispetto all’anamnesi
001_SLEISENGER_01_0003_0015.indd 14 24/02/18 15:44

Capitolo 1 Crescita cellulare e neoplasia 15
familiare di malattia. Nonostante questa limitata applicabilità clinica, l’identificazione di nuovi geni non precedentemente asso-ciati alla malattia aumenta la possibilità di nuovi approcci dia-gnostici e terapeutici. Un’altra evidenza derivata da questo tipo di studi è che tali varianti non sono necessariamente causali ma possono essere semplicemente associate, risultando in effetti in “linkage disequilibrium” con la variante di interesse.
La seconda ipotesi, definita come “malattia comune-variante rara” è basata sulla premessa che il rischio genetico di malattie come il cancro del colon-retto è principalmente determinato da un assetto eterogeneo di mutazioni rare o de novo. In molti studi, le varianti rare sono definite come quelle con frequenza inferiore all’1% nella popolazione generale. Confrontate con le varianti comuni, le varianti rare sono più inclini ad avere effetti più ampi a causa della capacità di purificare la selezione. Studi recenti, comunque, dimostrano che gran parte delle varianti rare sono comparse verosimilmente nell’arco degli ultimi 5000 anni e che tali varianti sono riconducibili all’espansione della popolazione e alla relativa minore eliminazione di queste varianti.83 I vantaggi degli studi sulle varianti rare sono che le varianti identificate risultano più frequentemente correlate direttamente con la malattia, tenuto conto della mancanza di linkage disequilibrium con altri alleli. Dato il maggior peso del loro effetto, queste varianti possono rivestire un ruolo cruciale anche nel processo clinico decisionale.
Sequenziamento dell’intero genoma e dell’esomaAlla luce della riduzione dei costi dei processi di sequenziamento del DNA, sussiste un discreto interesse nel correlare il profilo genomico completo delle neoplasie con la pratica clinica, con l’obiettivo di identificare terapie personalizzate per ogni indi-viduo. A oggi, due strategie sono attivamente seguite. La prima è il sequenziamento dell’intero genoma del tumore con l’obiet-tivo di conoscere anche le aree genomiche non codificanti, nel tentativo di individuare significati clinico-prognostici anche di queste sequenze genomiche. L’altro metodo si focalizza esclu-sivamente sull’esoma, la parte del genoma che codifica per le proteine. Sebbene comprenda solo l’1% del genoma, si ritiene che l’esoma contenga approssimativamente l’85% delle mutazioni associate alle malattie e il costo del suo sequenziamento risulta significativamente più contenuto. Numerose ricerche, comprese quelle del Cancer Genome Atlas Project, promosso dal National Cancer Institute, e dell’International Cancer Genome Consortium,
sono attualmente in corso, con il fine di catalogare le alterazioni alleliche in un ampio spettro di tumori.
BIBLIOGRAFIAPer i riferimenti bibliografici completi relativi a questo capitolo si rimanda al sito sleisenger.edizioniedra.it
2. Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012; 487:330-7.
11. Collado M, Blasco MA, Serrano M. Cellular senescence in cancer and aging. Cell 2007; 130:223-33.
22. Weisenberger DJ, Siegmund KD, Campan M, et al. CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nat Genet 2006; 38:787-93.
24. Gerlinger M, Rowan AJ, Horswell S, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med 2012; 366:883-92.
25. Nguyen LV, Vanner R, Dirks P, et al. Cancer stem cells: An evolving concept. Nat Rev Cancer 2012; 12:133-43.
33. Knudson AG Jr. Mutation and cancer: Statistical study of retinoblastoma. Proc Natl Acad Sci U S A 1971; 68:820-3.
58. Chan AT, Ogino S, Fuchs CS. Aspirin and the risk of colorectal cancer in relation to the expression of COX-2. N Engl J Med 2007; 356:2131-42.
60. Hutvagner G, McLachlan J, Pasquinelli AE, et al. A cellular function for the RNA-interference enzyme Dicer in the maturation of the let-7 small temporal RNA. Science 2001; 293:834-8.
69. Grivennikov SI, Wang K, Mucida D, et al. Adenoma-linked barrier defects and microbial products drive IL-23/IL-17-mediated tumour growth. Nature 2012; 491:254-8.
83. Tennessen JA, Bigham AW, O’Connor TD, et al. Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science 2012; 337:64-9.
001_SLEISENGER_01_0003_0015.indd 15 24/02/18 15:44