
BIOINFORMATICA STRUTTURALE - PARTE II - Polo A. … · 2018-12-22 · computazionali di simulazione...
8
BIOINFORMATICA STRUTTURALE - PARTE II Dinamica molecolare Con il termine dinamica molecolare (molecular dynamics, MD) si intende un insieme di tecniche computazionali di simulazione che, mediante l´integrazione delle equazioni del moto, permette di studiare la dinamica di evoluzione di un sistema fisico e chimico a livello atomico e molecolare. In particolare, lo studio della dinamica molecolare è fondamentale per comprendere appieno la funzione delle proteine e le loro interazioni. Le srutture proteiche depositate, derivate da studi di cristallizzazione e diffrazione a raggi X o da studi per NMR, sono paragonabili a fotografie o brevi filmati, ovvero non descrivono tutti i possibili stati, le numerose conformazioni che le proteine sono in grado di assumere. Le proteine sono, appunto, strutture dinamiche in grado di adattarsi a varie interazioni e di modulare le proprietà biochimiche e di legame in funzione delle transizioni strutturali. La dinamica molecolare è utilizzata sia nella ricerca di base che in progetti biotecnologici : Comprensione del folding : è possibile eseguire esperimenti di folding/ unfolding a partire da strutture note per chiarire gli aspetti cinetici (Lei & Duan, J Mol Biol. 370:196-206, 2007) Studio delle interazioni : la funzione cellulare di tutte le molecole - siano esse proteiche o di altro tipo - dipende in ultima analisi dalle interazioni (una molecola che non interagisce con alcun altro componente è irrilevante per il sistema). La dinamica molecolare può studiare le interazioni poiché esse sono mediate da meccanismi e forze che controllano anche le transizioni conformazionali, il folding ecc. E’ ben noto il meccanismo con cui il legame a siti specifici (allosterici) è in grado di modulare l’attività enzimatica; in realtà il legame di ligandi a una regione proteica si trasduce in altre parti della proteina, regolandone la conformazione e quindi le proprietà di legame ad altre proteine, al DNA ecc. Farmacogenomica : lo studio delle interazioni trova immediata applicazione biotecnologica nella farmacogenomica. Negli approcci tradizionali, nuovi farmaci sono sviluppati mediante modificazioni casuali e multiple di molecole già esistenti e successiva selezione dei principi attivi attraverso la caratterizzazione sperimentale dell’interazione con specifici recettori o molecole bersaglio. Grazie alle tecniche di studio delle strutture proteiche e di dinamica molecolare è invece possibile tagliare fortemente tempi e costi realizzando librerie di ligandi modificati in silico e prevedendone l’interazione. Comprensione dei meccanismi di voltage-gating : lo studio dei canali ionici, come quello del potassio KvAP (Cuello et al. 2004. Science 306:491-5), è fondamentale per il passaggio a fasi di ingegneria proteica volte a modificare la selettività ionica e rendere i canali mutanti in grado di trasportare ioni di
Transcript of BIOINFORMATICA STRUTTURALE - PARTE II - Polo A. … · 2018-12-22 · computazionali di simulazione...

BIOINFORMATICA STRUTTURALE - PARTE II Dinamica molecolare Con il termine dinamica molecolare (molecular dynamics, MD) si intende un insieme di tecniche computazionali di simulazione che, mediante l´integrazione delle equazioni del moto, permette di studiare la dinamica di evoluzione di un sistema fisico e chimico a livello atomico e molecolare. In particolare, lo studio della dinamica molecolare è fondamentale per comprendere appieno la funzione delle proteine e le loro interazioni. Le srutture proteiche depositate, derivate da studi di cristallizzazione e diffrazione a raggi X o da studi per NMR, sono paragonabili a fotografie o brevi filmati, ovvero non descrivono tutti i possibili stati, le numerose conformazioni che le proteine sono in grado di assumere. Le proteine sono, appunto, strutture dinamiche in grado di adattarsi a varie interazioni e di modulare le proprietà biochimiche e di legame in funzione delle transizioni strutturali. La dinamica molecolare è utilizzata sia nella ricerca di base che in progetti biotecnologici : Comprensione del folding: è possibile eseguire esperimenti di folding/ unfolding a partire da strutture note per chiarire gli aspetti cinetici (Lei & Duan, J Mol Biol. 370:196-206, 2007) Studio delle interazioni: la funzione cellulare di tutte le molecole - siano esse proteiche o di altro tipo - dipende in ultima analisi dalle interazioni (una molecola che non interagisce con alcun altro componente è irrilevante per il sistema). La dinamica molecolare può studiare le interazioni poiché esse sono mediate da meccanismi e forze che controllano anche le transizioni conformazionali, il folding ecc. E’ ben noto il meccanismo con cui il legame a siti specifici (allosterici) è in grado di modulare l’attività enzimatica; in realtà il legame di ligandi a una regione proteica si trasduce in altre parti della proteina, regolandone la conformazione e quindi le proprietà di legame ad altre proteine, al DNA ecc. Farmacogenomica: lo studio delle interazioni trova immediata applicazione biotecnologica nella farmacogenomica. Negli approcci tradizionali, nuovi farmaci sono sviluppati mediante modificazioni casuali e multiple di molecole già esistenti e successiva selezione dei principi attivi attraverso la caratterizzazione sperimentale dell’interazione con specifici recettori o molecole bersaglio. Grazie alle tecniche di studio delle strutture proteiche e di dinamica molecolare è invece possibile tagliare fortemente tempi e costi realizzando librerie di ligandi modificati in silico e prevedendone l’interazione. Comprensione dei meccanismi di voltage-gating: lo studio dei canali ionici, come quello del potassio KvAP (Cuello et al. 2004. Science 306:491-5), è fondamentale per il passaggio a fasi di ingegneria proteica volte a modificare la selettività ionica e rendere i canali mutanti in grado di trasportare ioni di

metalli pesanti. Organismi in cui i canali sono stati mutati in tal senso possono essere utilizzati in programmi di decontaminazione ambientale (bioremediation e phytoremediation). Effetto di mutazioni: qui la dinamica molecolare viene impiegata ad esempio per lo studio del folding anomalo che caratterizza determinate proteine patogene come il prione, o nello studio di patologie multifattoriali dove un errato ripiegamento delle proteine che interagiscono con gli acidi nucleici è un fattore chiave nell’insorgenza di queste malattie (Barducci A et al., Biophys J 88,1334, 2005).
Nella dinamica molecolare si associa all’energia potenziale degli atomi della proteina una funzione che descrive una superficie o Potential Energy Surface (PES). (maggiori dettagli nella sezione – fuori programma – di approfondimento). I metodi per il calcolo dell’energia possono essere metodi quantomeccanici, che tengono conto di posizione e velocità di nuclei ed elettroni e metodi classici in cui sono considerati solo i nuclei, Tra i metodi classici, la meccanica molecolare si estrinseca nel force field, cioè un potenziale che descrive le caratteristiche energetiche della molecola (maggiori dettagli nella sezione – fuori programma – di approfondimento). A seconda delle esigenze, la dinamica può essere implementata in differenti simulazioni di fase sovente:
- Solvente esplicito --> è necessaria una simulazione più accurata ed è quindi simulata l’aggiunta di molecole d’acqua al sistema. In caso di proteine con cariche nette vengono aggiunti controioni per neutralizzare il sistema. Questo metodo è più accurato ma anche più dispendioso computazionalmente. - Solvente implicito --> la presenza del solvente è simulato da una “costante dielettrica efficiente”. In questo caso il metodo è meno accurato ma il costo computazionale è notevolmente ridotto rispetto alla trattazione con solvente esplicito.
La fase di minimizzazione energetica è necessaria per eliminare gli atom clashes; in pratica il sistema è virtualmente portato alla temperatura desiderata per rientropizzare e consentire nuovamente il raggiungimento di minimi energetici. Dopo la minimizzazione avviene uno step di equilibrazione del sistema, in cui si cerca di mantenere il sistema stesso alla temperatura desiderata. Successivamente alla fase di equilibrazione possono prendere via i cicli produttivi (production runs). Dopo un esperimento di dinamica molecolare, il parametro RMSD precedentemente introdotto è utilizzato come indicatore del raggiungimento del folding (se la distanza compresa rispetto alla struttura iniziale è tra 3 e 4°). Il processo di folding è tipicamente accompagnato da un decremento significativo della SASA (Solvent Accessible Surface Area).
(maggiori dettagli nella sezione – fuori programma – di approfondimento).
Predizione di interazioni intramolecolari Per “assemblare” i vari elementi di struttura secondaria in un contesto 3D non modellizzabile più o meno facilmente grazie ad un templato può essere utile considerare le interazioni intramolecolari a medio e lungo raggio. La predizione di contatti è fatta “a posteriori”, ovvero considerando l’effetto di mutazioni in coppia, compensative per il mantenimento della funzione. Ad esempio, nell’allineamento:
Nella posizione contornata dal box blu, tutte le sequenze hanno un residuo aromatico grande (triptofano, W, tirosina, Y, fenilalanina, F) tranne una (che mostra una glicina, G). Nella posizione contornata dal box rosso, quasi tutte le sequenze hanno un residuo piccolo (G o alanina, A), ma la

quinta dall’alto ha un residuo grande (W). In questa sequenza l'ordine grande-piccolo per i residui nelle posizioni contornate dai due box è invertito rispetto alle altre cinque sequenze; se è noto che per le sei proteine la funzione è conservata, allora la doppia mutazione è tollerata, suggerendo che i residui nelle due posizioni siano in contatto e formino coppie di ingombro sterico equivalente, ovvero “grande + piccolo” oppure “piccolo + grande”. Invece, nell’allineamento:
il contatto di coppia è tra residui di carica opposta, negativa in posizione N-ter (box blu) e positiva in C-ter (box rosso), tranne che in una sequenza, che ha le posizioni invertite ma carica complessiva immutata (poiché -1 +1 oppure +1 –1 è comunque pari a zero). Ci sono, tuttavia, limiti in questo tipo d'analisi; talora è difficile riconoscere l’effetto di compensazione poichè non sempre è mediato da sostituzioni compensative di altri residui. Infatti, il backbone proteico ha una certa elasticità ed, entro certi limiti, può consentire un certo "accomodamento" dei residui. Inoltre, non necessariamente la compensazione “a coppie” avviene tra singoli residui: la compensazione può riguardare gruppi di residui. Ad esempio, nell’allineamento:
I residui in rosso nei riquadri sono complessivamente aromatici o idrofobici ed il loro ingombro sterico totale è più o meno costante. Ad esempio, nel gruppo N-ter (box blu) tutte le sequenze hanno due residui grandi (W, F o Y) ed uno piccolo (G o A) ma una di esse ha tre residui di grandezza intermedia (V); nella posizione C-ter (box rosso) tutte hanno un residuo V e due residui piccoli. Comprendere appieno quanto siano importanti queste interazioni a breve, medio e lungo raggio è ha rilevanza funzionale e, in ultima analisi, anche biotecnologica, poichè determina il successo o meno dei progetti di ingegneria proteica. Infatti, quando si progettano mutazioni per modificare nel senso voluto le proprietà di una proteina, non si deve dimenticare che ciascuno degli aminoacidi presenti nella proteina wild type fa parte di un network di interazioni con numerosi altri residui della stessa catena polipeptidica o, talora, di residui di altri monomeri o interattori. Quando sostituiamo uno di tali residui con un altro, per modificare la proprietà che ci interessa, dobbiamo pensare al fatto che, come in un domino intramolecolare, tutti i residui che prima interagivano con l'aminoacido originale, ora si trovano un residuo diverso. Conseguentemente, alcune interazioni possono essere perse, altre alterate, altre stabilite, spostando legami idrogeno, idrofobici e ponti salini, con i tipici riflessi negativi su stabilità e solubilità mostrati nei progetti di rational design. Una progettazione più evoluta deve quindi non solo guardare alla modifica, ma anche alla ristabilizzazione degli equilibri perduti, altrimenti il lavoro svolto per ottenere un enzima più processivo o capace di catalisi modificata è vanificato dal fatto che lo stesso, pur più attivo potenzialmente, o dotato di nuova attività, si mostra inutilizzabile in quanto insolubile o rapidamente degradato.

Simulazioni di docking Se la struttura di due proteine è conosciuta ed è noto che interagiscono, non è sufficiente comparare le due strutture per dedurre le regioni di interazione. Infatti, la struttura di una proteina può cambiare se ad essa è complessato un ligando proteico o di altro tipo (docking):
Nella predizione di docking può essere di aiuto l’identificazione di “mutazioni correlate” tra allineamenti di proteine per le quali sia noto che formano complessi.Il docking non riguarda solo la formazione di complessi tra due o più proteine ma anche l’interazione con acidi nucleici o piccoli ligandi ionici o lipidici. Il complesso tra proteine procariotiche spesso è simile alla struttura 3D di domini omologhi in una proteina eucariotica più grande. Nell'esempio in figura, i complessi procariotico (a sinistra) ed eucariotico (a destra) hanno struttura simile, ma nel procariote le subunità sono quattro (A, B, C, D) mentre nell'eucariote sono solo due: la proteina A, che è conservata ed una singola proteina E composta da tre domini corrispondenti alle proteine procariotiche B, C e D):
Le predizioni di docking sono fondamentali nell'ingegneria proteica e nello sviluppo di farmaci, poichè ad esempio permette di predire se la proteina mutante progettata sarà in grado di legare meglio o peggio un dato ligando, oppure se una molecola biomimetica sarà in grado di legare un recettore, e così via. Esistono differenti piattaforme che consentono di svolgere simulazioni di docking da remoto, alcune tra proteine, altre tra proteine e piccoli ligandi.
APPROFONDIMENTO (OPZIONALE)
(per chi trovi la Bioinformatica Strutturale argomento preferito - o tra i preferiti - del corso)
Vediamo quali sono gli “ingredienti” per effettuare una dinamica molecolare: • Spazio conformazionale (PES): Gli atomi costituenti la proteina possiedono una Epot che
dipende solo dalle coordinate dei suoi atomi. All’energia potenziale è associata una funzione che descrive una superficie, detta PES (Potential Energy Surface), caratterizzata da elevata dimensionalità [(3N-6)+1] e rappresentabile solo quando le dimensioni sono N<3. La PES rappresenta lo spazio conformazionale della proteina e non è piatto, ma viene rappresentato col modello ad imbuto, in cui l’energia libera fornisce la profondità dell’imbuto mentre la larghezza dell’imbuto stesso è rappresentata dall’entropia.
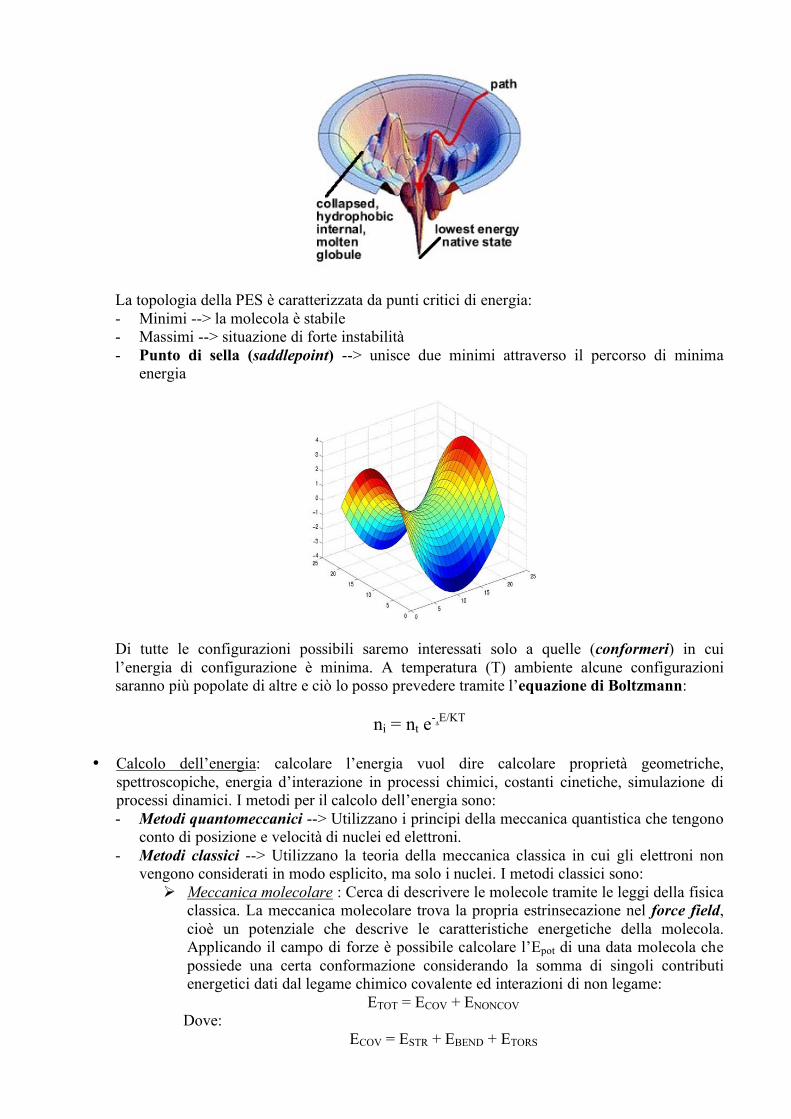
La topologia della PES è caratterizzata da punti critici di energia: - Minimi --> la molecola è stabile - Massimi --> situazione di forte instabilità - Punto di sella (saddlepoint) --> unisce due minimi attraverso il percorso di minima
energia
Di tutte le configurazioni possibili saremo interessati solo a quelle (conformeri) in cui l’energia di configurazione è minima. A temperatura (T) ambiente alcune configurazioni saranno più popolate di altre e ciò lo posso prevedere tramite l’equazione di Boltzmann:
ni = nt e-ΔE/KT
• Calcolo dell’energia: calcolare l’energia vuol dire calcolare proprietà geometriche,
spettroscopiche, energia d’interazione in processi chimici, costanti cinetiche, simulazione di processi dinamici. I metodi per il calcolo dell’energia sono: - Metodi quantomeccanici --> Utilizzano i principi della meccanica quantistica che tengono
conto di posizione e velocità di nuclei ed elettroni. - Metodi classici --> Utilizzano la teoria della meccanica classica in cui gli elettroni non
vengono considerati in modo esplicito, ma solo i nuclei. I metodi classici sono: Meccanica molecolare : Cerca di descrivere le molecole tramite le leggi della fisica
classica. La meccanica molecolare trova la propria estrinsecazione nel force field, cioè un potenziale che descrive le caratteristiche energetiche della molecola. Applicando il campo di forze è possibile calcolare l’Epot di una data molecola che possiede una certa conformazione considerando la somma di singoli contributi energetici dati dal legame chimico covalente ed interazioni di non legame:
ETOT = ECOV + ENONCOV Dove:
ECOV = ESTR + EBEND + ETORS

ESTR = ESTRETCHING = funzione per l’allungamento di un legame tra due atomi EBEND = EBENDING = energia necessaria per deformare un angolo formato da tre atomi ABC quando ci sia un legame tra A e B e tra B e C ETORS = ETORSION = energia di torsione attorno ad un legame per formare un nuovo angolo diedro.
ENONCOV = EEL + EVdW EEL = energia elettrostatica, calcolata utilizzando il potenziale di Coulomb EVdW = termine legato alle forze di Van der Waals, calcolato col potenziale di Lennard-Jones.
Dinamica Molecolare: nella dinamica molecolare si adopera il force field al fine di valutare il comportamento dinamico di una molecola, dunque si vede come si muovono gli atomi in un determinato tempo. La proteina deve esplorare il bacino di attrazione del minimo globale o di altri minimi. Se T≠ 0 si parla di energia cinetica e di energia potenziale:
ETOT = EK + EPOT Dove: EK = Energia cinetica = ½ mv2 = 3/2 KBT
Fornendo energia alla molecola, questa si muoverà lungo la PES, descrivendo una traiettoria. Considerando il sistema proteina:
abbiamo tante particelle che compongono il sistema le particelle interagiscono e la forza che agisce su un atomo cambia ad ogni δt
perchè gli atomi si muovono e cambiano posizione. Se le condizioni iniziali del moto a cui è sottoposta la particella sono note, possono essere scritte le leggi del moto. Quindi ad un certo istante t si sa come calcolare l’EPOT, ma nel t successivo la configurazione è un po’ cambiata perchè ci sono delle forze che agiscono sull’atomo. La legge che governa il moto dei vari atomi è:
F = ma
Lo scopo è risolvere l’equazione del moto per ricavare la posizione r(t) e la velocità v(t). Visto che ad ogni istante tutto cambia, non è possibile integrare l’equazione del moto ma si segue l’evoluzione della traiettoria ad ogni δt. In che modo? La traiettoria è continua ma si procede per intervalli discretizzati. Applicando la regola del trapezio, più piccolo è il trapezio meglio approssimo l’integrale della curva. Quindi, riassumendo, ad ogni passo vengono calcolati:
Forze come derivate di energia potenziale rispetto alle coordinate atomiche --> Fi(t) = -∂EPOT/∂T
Aggiornamento delle velocità e delle coordinate atomiche mediante le forze precedentemente ottenute.
Una dinamica molecolare può essere condotta in differenti ensembles. Con questo termine si suole indicare l’idealizzazione di un gran numero di copie del sistema considerate all’istante, ognuna delle quali rappresenta un possibile stato in cui il sistema reale potrebbe essere. Nella molecola le particelle calde dovranno cedere energia cinetica alle particelle fredde e allora compariranno delle forze: c’è un travaso da una parte all’altra di energia cinetica e potenziale. Si può lavorare in:
NVE ensemble (microcanonical ensemble) --> E’ un sistema isolato in cui il numero di particelle, velocità ed energia totale vengono conservati. NVE corrisponde ad un processo adiabatico in cui non c’è scambio di calore. Sfortunatamente, il microcanonical ensemble non corrisponde alle condizioni alle quali avviene la maggior parte degli esperimenti. Infatti, se lavoro ad ETOT costante, l’energia cinetica istantanea varia e cambia la T istantanea. Non ha

senso portare la proteina in condizioni di alta T; se per caso siamo andati in un punto in cui le forze sono grandi e la PES s’impenna, le velocità aggiornate saranno grandi e vado a finire in un regime ad alta T.
NVT ensemble (canonical ensemble) --> Abbandonata l’idea di lavorare ad energia costante per i motivi esposti precedentemente, si decide di lavorare a temperatura costante: in questo modo l’energia cinetica sarà costante ma varierà l’energia potenziale, con la conseguenza che l’energia totale cambia. Questo è l’approccio più fisiologico in quanto questo non è un sistema isolato; per mantenere costante la T bisogna invocare un sistema di scambio di calore tra il mio sistema e l’universo (bagno termostatico). Se l’aumento della T è molto grande, il sistema impiegherà molto più tempo a ritermalizzarsi: ciò vuol dire che la mia proteina rischia di stare per troppo tempo ad alta T. per mantenere costante la T bisogna accoppiare il sistema proteina con un bagno termostatico esterno, la cui T è circa costante ed è controllata da un termostato. L’ensemble NVT è fondamentale nello studio di protein unfolding che richiede un controllo preciso della T.
NPT ensemble --> In questo caso P e T vengono mantenuti costanti. Una volta scelto l’ensemble opportuno, è necessario decidere se effettuare la dinamica in solvente esplicito o implicito: Solvente esplicito --> Vengono aggiunte molecole d’acqua al sistema
(solvation box) e la distanza che intercorre tra gli atomi della proteina e lo spigolo della box dovrebbe essere compreso tra gli 8 e i 10 A. In caso di proteine con cariche nette vengono aggiunti controioni per neutralizzare il sistema.
Solvente implicito --> Consiste nell’inclusione di una costante elettrostatica nel termine elettrostatico dell’energia potenziale (costante dielettrica efficiente). Le molecole di solvente sono implicitamente considerate modificando opportunamente le interazioni tra i nuclei come se essi subissero l'effetto ``medio'' cioe' statistico della presenza di un gran numero di molecole di solvente. In questo caso il costo computazionale è notevolmente ridotto rispetto alla trattazione con solvente esplicito.
A questo punto avviene una fase di minimizzazione energetica. Questa fase è necessaria per eliminare quelli che vengono definiti come atom clashes. Solitamente si effettuano alcuni cicli di steepest descent o steepest descent+conjugate gradients ed il sistema viene portato alla temperatura desiderata. Dopo la minimizzazione avviene uno step di equilibrazione del sistema, in cui si cerca di mantenere il sistema stesso alla temperatura desiderata. In questa fase la temperatura viene costantemente monitorata e se avvengono variazioni di un tot K (ad es. ±10K) rispetto alla T preimpostata, le velocità vengono scalate per consentire la ritermalizzazione alla T voluta. Successivamente alla fase di equilibrazione (della durata di circa 20ps) possono prendere via i cicli produttivi (production runs). Dall’analisi della traiettoria (o dalle traiettorie) ottenute da un esperimento di dinamica molecolare si ottengono importanti informazioni:
RMSD: Come già visto precedentemente,rappresenta la vicinanza, in termini strutturali, alla struttura nativa. Viene solitamente considerato l’RMSD dei Cα Una distanza compresa tra 3 e 4A rispetto alla struttura iniziale è generalmente usata come indicatore del raggiungimento del folding.
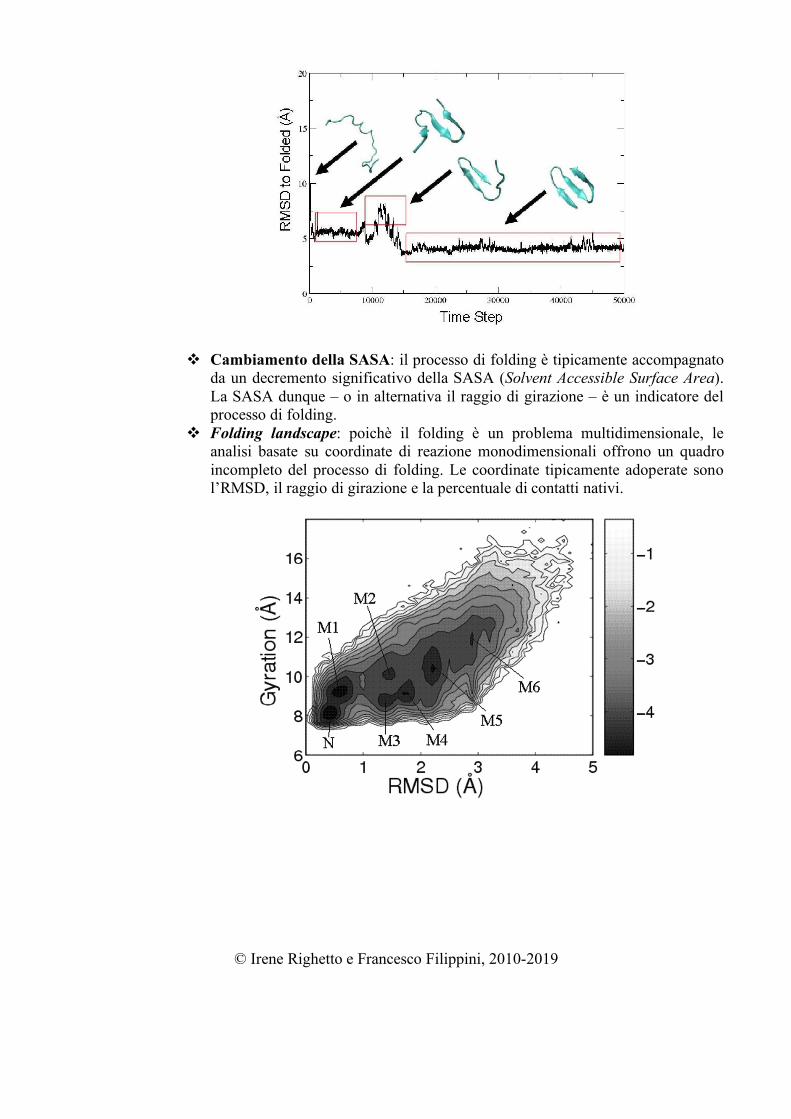
Cambiamento della SASA: il processo di folding è tipicamente accompagnato
da un decremento significativo della SASA (Solvent Accessible Surface Area). La SASA dunque – o in alternativa il raggio di girazione – è un indicatore del processo di folding.
Folding landscape: poichè il folding è un problema multidimensionale, le analisi basate su coordinate di reazione monodimensionali offrono un quadro incompleto del processo di folding. Le coordinate tipicamente adoperate sono l’RMSD, il raggio di girazione e la percentuale di contatti nativi.
© Irene Righetto e Francesco Filippini, 2010-2019


















