
Anomalie Cromosomiche Di Numero
45
Anomalie cromosomiche - 1 • Bilanciate: nella maggioranza dei casi non sono correlate ad un fenotipo anomalo • Sbilanciate: sono correlate ad un fenotipo anomalo (malformazioni e/o ritardo mentale)
Transcript of Anomalie Cromosomiche Di Numero

Anomalie cromosomiche - 1
• Bilanciate: nella maggioranza dei casi non sono correlate ad un fenotipo anomalo • Sbilanciate: sono correlate ad un fenotipo anomalo (malformazioni e/o ritardo mentale)
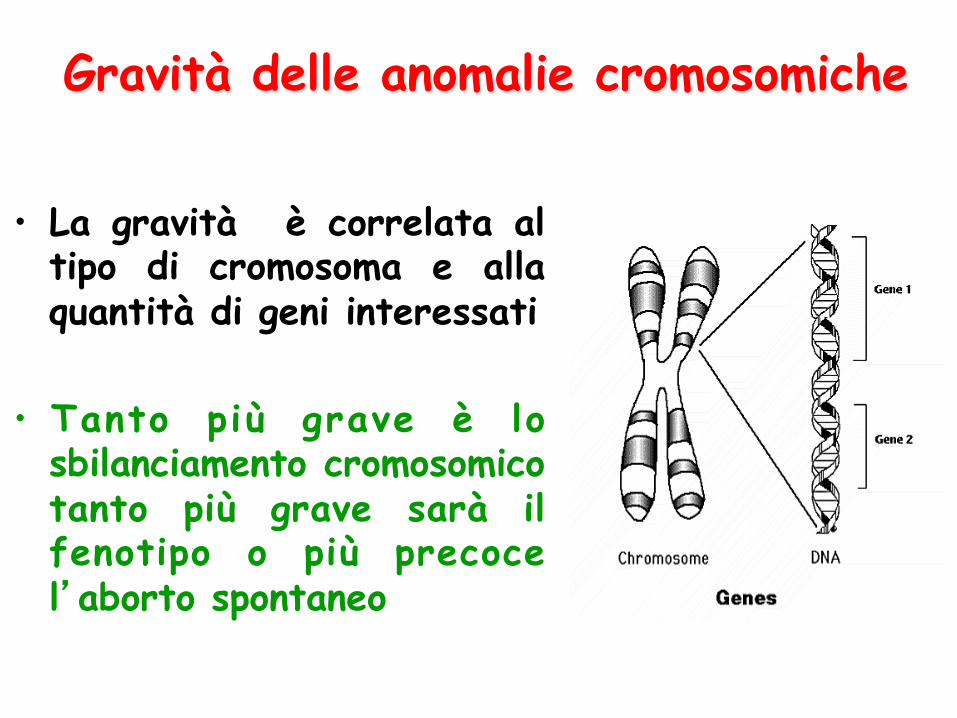
Gravità delle anomalie cromosomiche
• La gravità è correlata al tipo di cromosoma e alla quantità di geni interessati
• Tanto più grave è lo sbilanciamento cromosomico tanto più grave sarà il fenotipo o più precoce l’aborto spontaneo

ANOMALIE CROMOSOMICHE - 2
del numero della struttura
• poliploidie (triploidie, tetraploidie) • aneuploidie (trisomie, monosomie) • mosaici • marcatori sovrannumerari
Bilanciate • traslocazioni (reciproche e robertsoniane) • inversioni (paracentriche e pericentriche)
Sbilanciate • delezioni • duplicazioni • isocromosomi • cromosomi ad anello
Sbilanciate

Le anomalie cromosomiche rappresentano la
patologia più frequente del concepimento e,
nonostante la fortissima selezione naturale
mediante l’aborto spontaneo, la loro frequenza
alla nascita è elevata.
Anomalie cromosomiche - 3

Epidemiologia delle anomalie cromosomiche
L’alta incidenza di anomalie cromosomiche suggerisce che questa patologia, riscontrata uniformemente in tutte le popolazioni, sia una caratteristica della riproduzione umana e che sia poco influenzata da condizioni etniche e geografiche.

NATI VIVI La frequenza delle alterazioni del cariotipo alla nascita, in
assenza di diagnosi prenatale, è circa dell’1%. Anomalie cromosomiche Frequenza (%)
Numeriche autosomiche Numeriche cromosomi del sesso
0,17 Maschi 0,12 Femmine 0,07
Strutturali bilanciate sbilanciate
0,52 0,06
Totali 0,91

ANOMALIE CROMOSOMICHE DI NUMERO
• La POLIPLOIDIA è l a c o s t i t u z i o n e cromosomica di una cellula, o di un organismo, che possiede tre o più assetti cromosomici n.
• Le anomalie di numero di singoli cromosomi
sono chiamate ANEUPLOIDIE.
La presenza di tre copie dello stesso cromosoma è chiamata TRISOMIA, di una sola copia MONOSOMIA.

Ch.1 Ch.2 Ch.3 Ch.4 Ch.5
…… ASSETTO DIPLOIDE CORRETTO
MONOPLOIDIA (aploide)
POLIPLOIDIA
(triploidia)
MONOSOMIA
TRISOMIA
NULLISOMIA

Poliploidia
• Si definiscono poliploidi le cellule con un numero di cromosomi corrispondente ad un multiplo esatto del corredo aploide (n)
• Triploidia (3n= 69 cromosomi) • Tetraploidia (4n= 92 cromosomi)
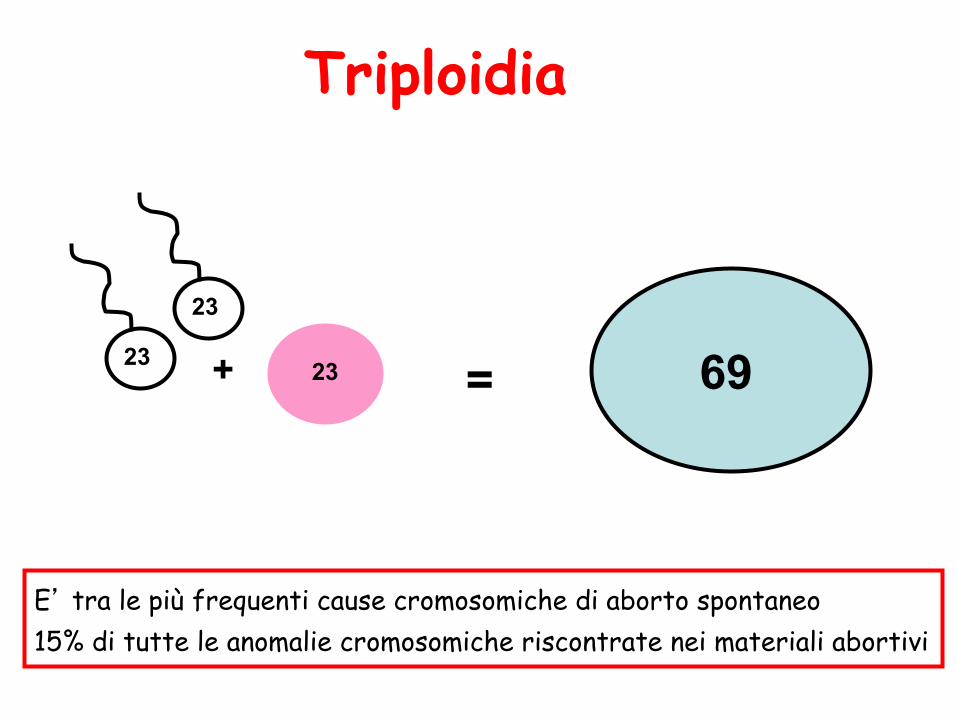
Triploidia
23 23 69 + = 23
E’ tra le più frequenti cause cromosomiche di aborto spontaneo 15% di tutte le anomalie cromosomiche riscontrate nei materiali abortivi
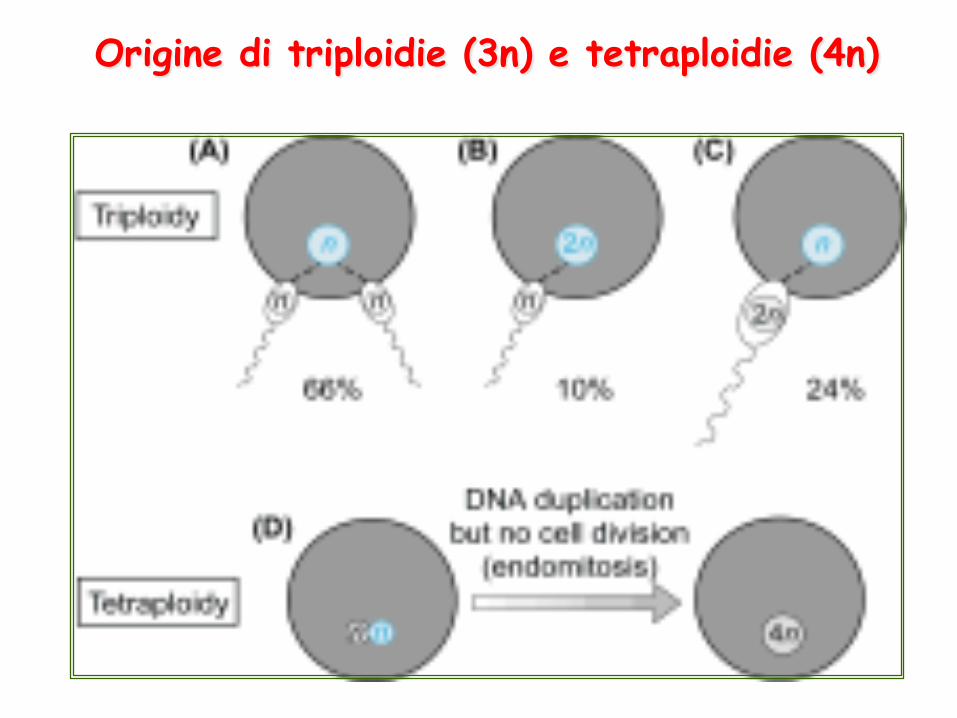
Origine di triploidie (3n) e tetraploidie (4n)

Aneuploidie • Si definiscono aneuploidi le cellule con cromosomi
in più o in meno, ma non equivalenti a un multiplo esatto del corredo aploide
– Trisomie – Monosomie – Marcatori sovrannumerari
L’aneuploidia è i principale fattore eziologico alla base di un alterato sviluppo fetale e, nella maggior parte dei casi, esita in aborto

• Frequenza nei nati vivi 0,36%
• Il principale meccanismo all’origine è la non-disgiunzione cromosomica durante la meiosi
• Rischio correlato all’età materna

Aneuploidie sono causate da: • Non-disgiunzione
– Mancata separazione degli omologhi in anafase I della meiosi
– Mancata separazione dei cromatidi alla meiosi II
• Ritardo (lag) anafasico – E’ causato da un ritardo nella migrazione del
cromosoma/cromatide durante l’anafase. – Dà origine ad una cellula figlia in cui manca un
cromosoma o un cromatidio.
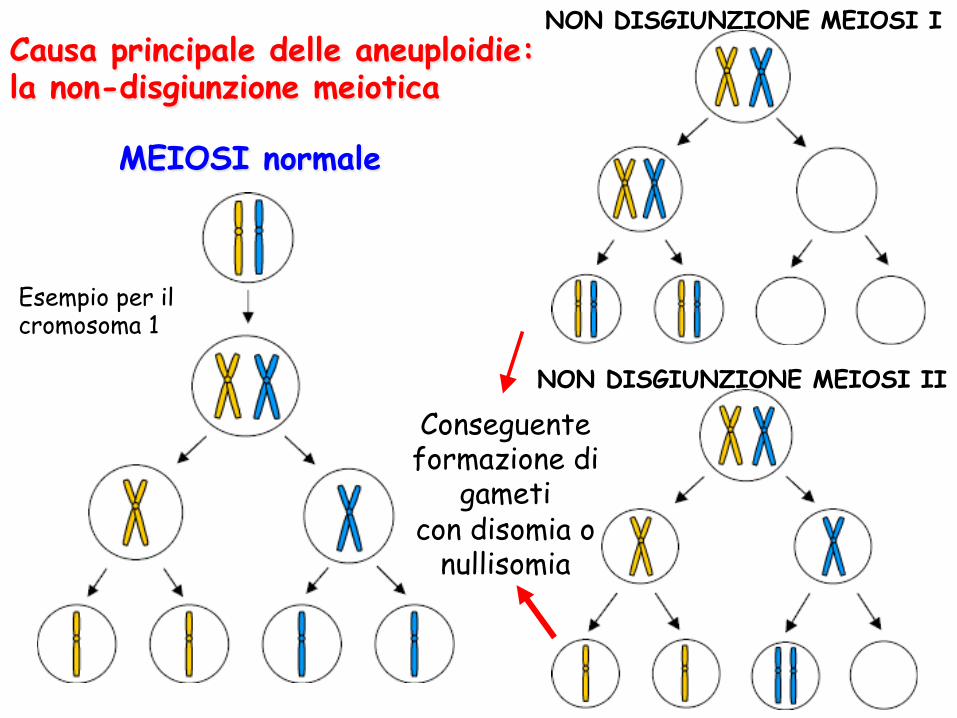
MEIOSI normale
NON DISGIUNZIONE MEIOSI I
NON DISGIUNZIONE MEIOSI II
Causa principale delle aneuploidie: la non-disgiunzione meiotica
Esempio per il cromosoma 1
Conseguente formazione di
gameti con disomia o
nullisomia
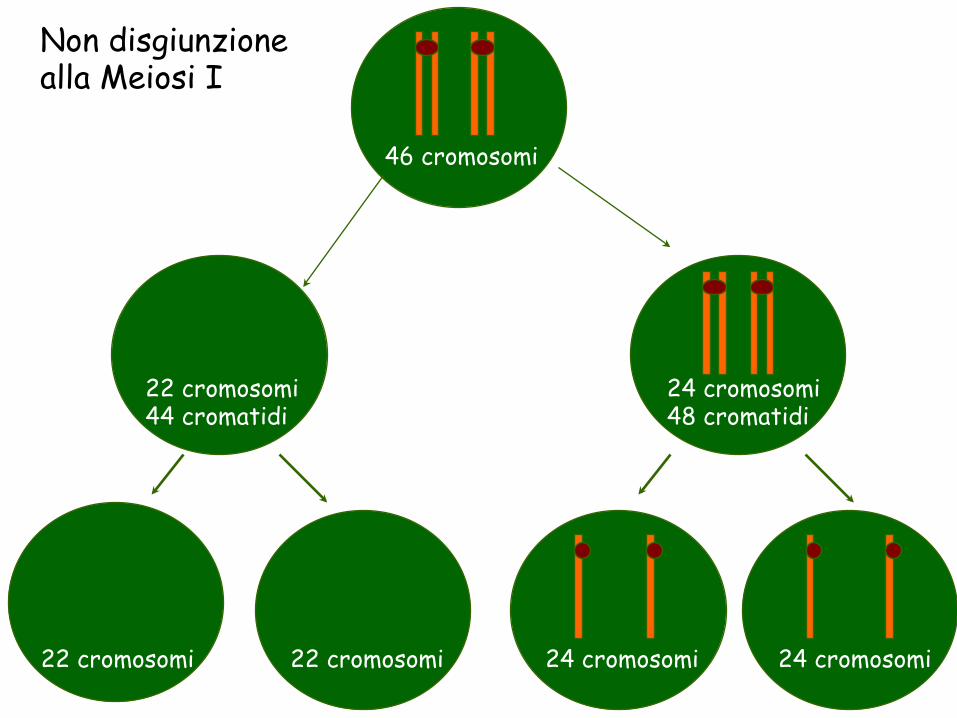
46 cromosomi
24 cromosomi 48 cromatidi
22 cromosomi 44 cromatidi
22 cromosomi 22 cromosomi 24 cromosomi 24 cromosomi
Non disgiunzione alla Meiosi I
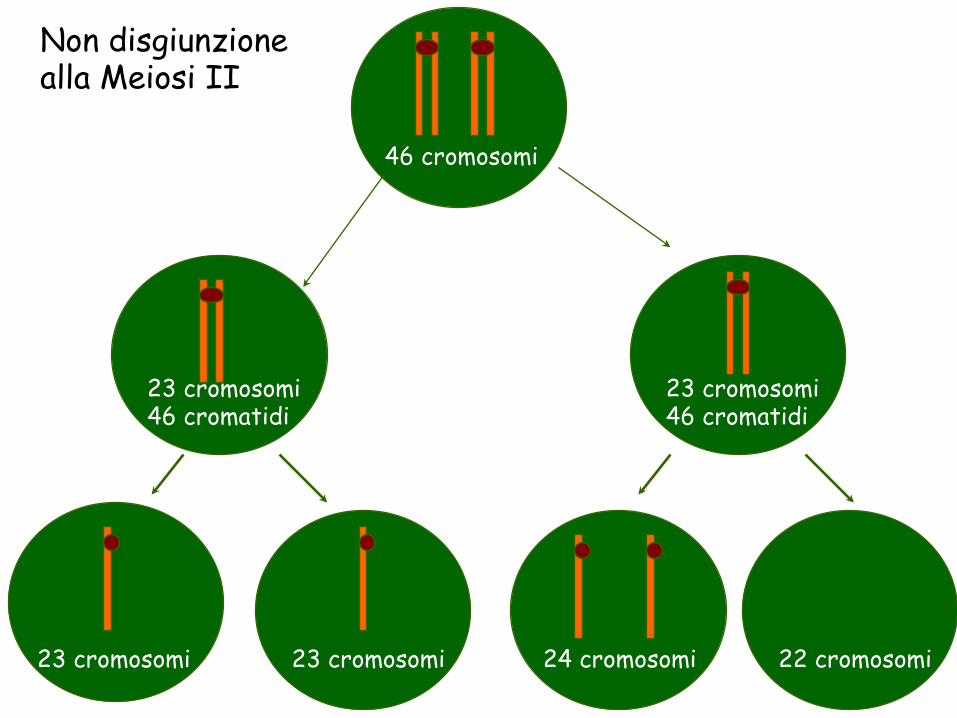
46 cromosomi
23 cromosomi 46 cromatidi
23 cromosomi 46 cromatidi
23 cromosomi 23 cromosomi 24 cromosomi 22 cromosomi
Non disgiunzione alla Meiosi II

Condizioni causate da non-disgiunzione Sindrome di Down = Trisomia 21 3 copie di cromosomi 21 per un totale di 47 cromosomi
+ =
Sindrome di Turner = Monosomia X 1 solo cromosoma sessuale (X) per un totale di 45 cromosomi
+ =
Sindrome di Klinefelter = XXY 1 cromosoma X in più
+ =
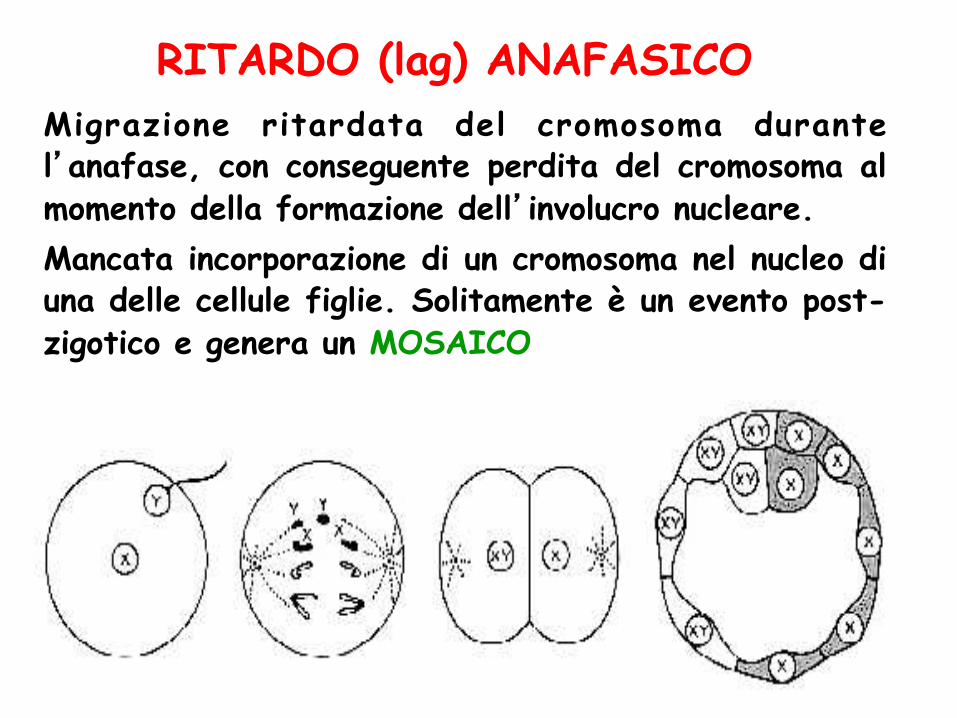
Migrazione ritardata del cromosoma durante l’anafase, con conseguente perdita del cromosoma al momento della formazione dell’involucro nucleare. Mancata incorporazione di un cromosoma nel nucleo di una delle cellule figlie. Solitamente è un evento post-zigotico e genera un MOSAICO
RITARDO (lag) ANAFASICO

MOSAICO Presenza in uno stesso individuo di due o più linee cellulari geneticamente diverse che derivano da un unico zigote
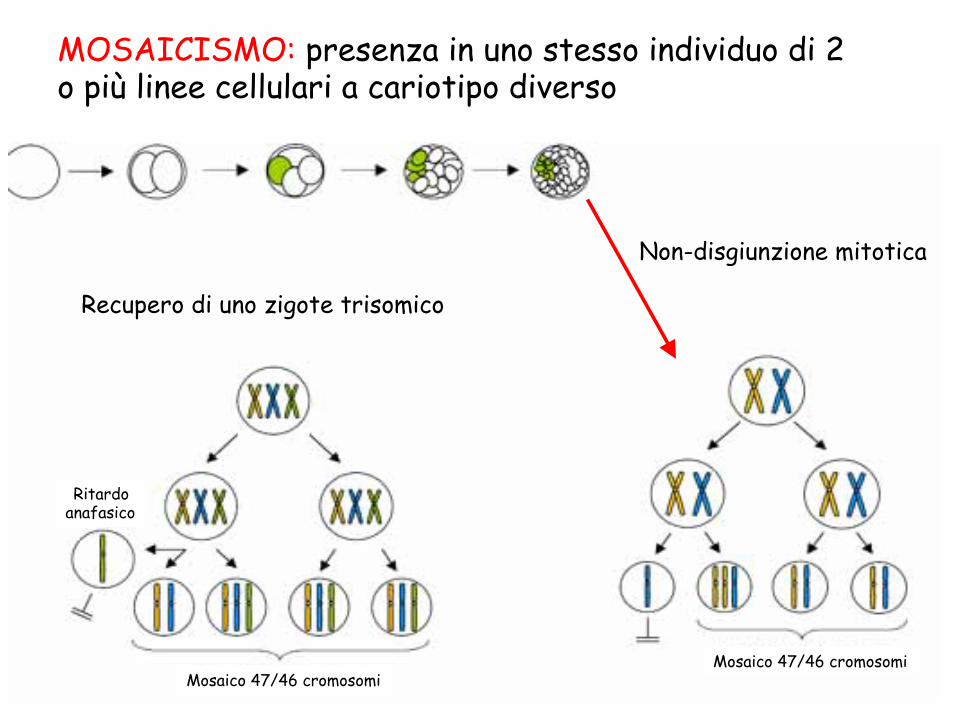
MOSAICISMO: presenza in uno stesso individuo di 2 o più linee cellulari a cariotipo diverso
Ritardo anafasico
Mosaico 47/46 cromosomi Mosaico 47/46 cromosomi
Recupero di uno zigote trisomico
Non-disgiunzione mitotica
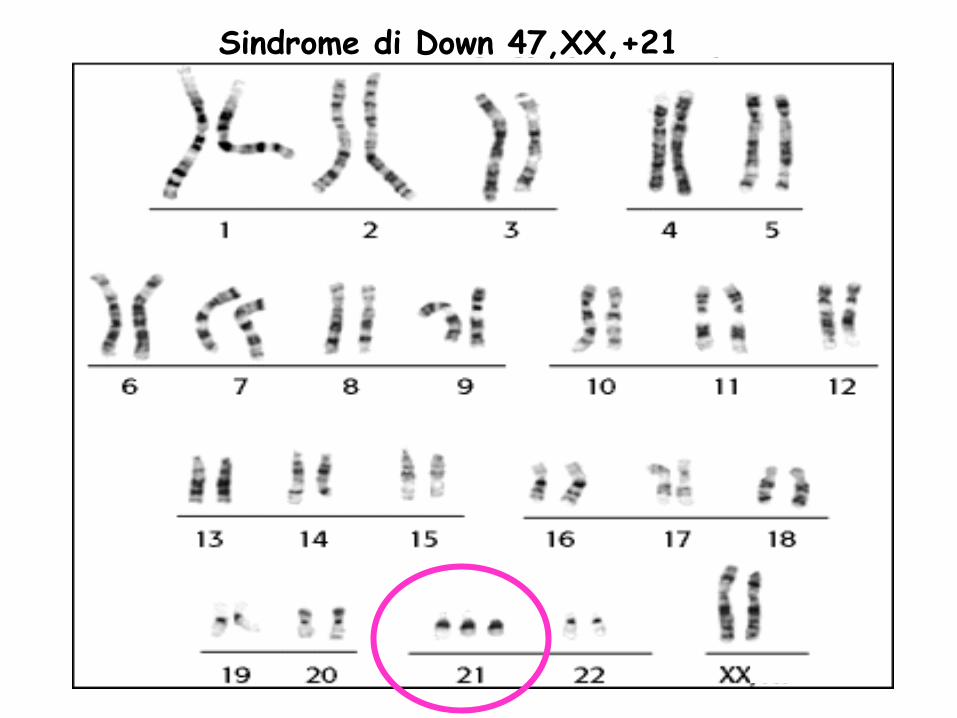
Sindrome di Down 47,XX,+21

SINDROME DI DOWN
• E’ una delle anomalie cromosomiche più frequenti (1:700 nati vivi). Al concepimento la frequenza è molto più elevata, 2/3 dei +21 vengono abortiti spontaneamente e il 20% nascono morti.
• Tale frequenza risulta costante nelle diverse popolazioni del mondo.
• L’unico fattore che ne modifica la prevalenza è rappresentato dalla diffusione della diagnosi prenatale.
• E’ la più frequente condizione genetica associata a ritardo psicomotorio.
• L’incidenza aumenta con l’aumentare dell’età materna e il 95% delle trisomie sono causate da errori di non-disgiunzione durante la meiosi materna.
• Nel 5% casi la trisomia è trasmessa da un genitore portatore di una traslocazione robertsoniana coinvolgente il cromosoma 21.

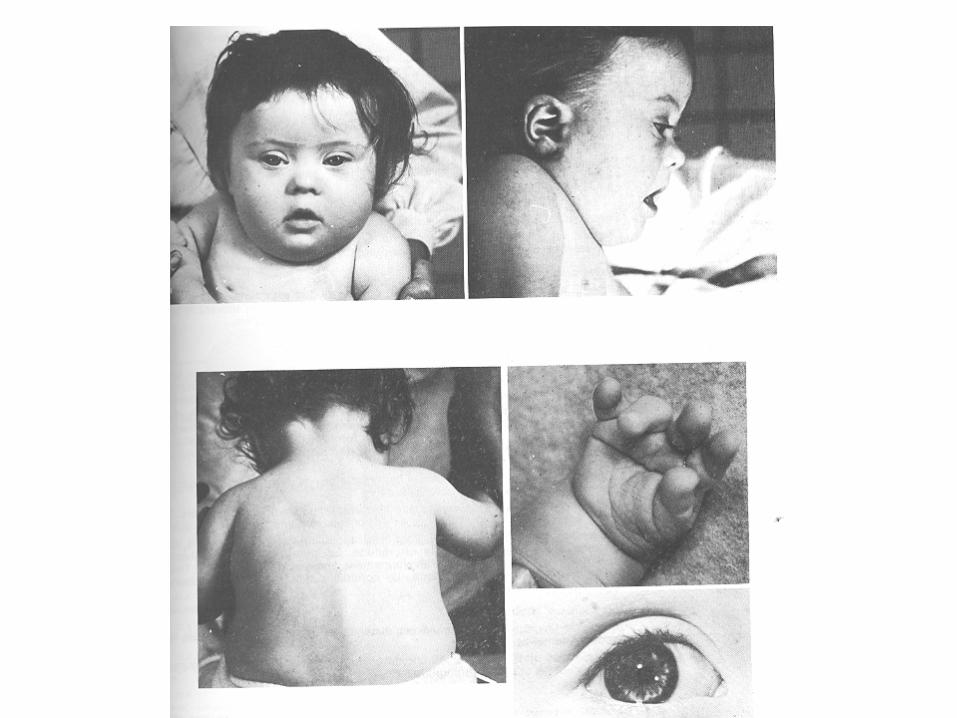
Caratteristiche fenotipiche della sindrome di Down: • ipotonia • mongolismo (corpo basso e tozzo e collo grosso)
• viso rotondo e piatto • macroglossia • taglio palpebrale obliquo • orecchie piccole • nuca piatta • cardiopatie congenite • microcefalia • ritardo psico-motorio
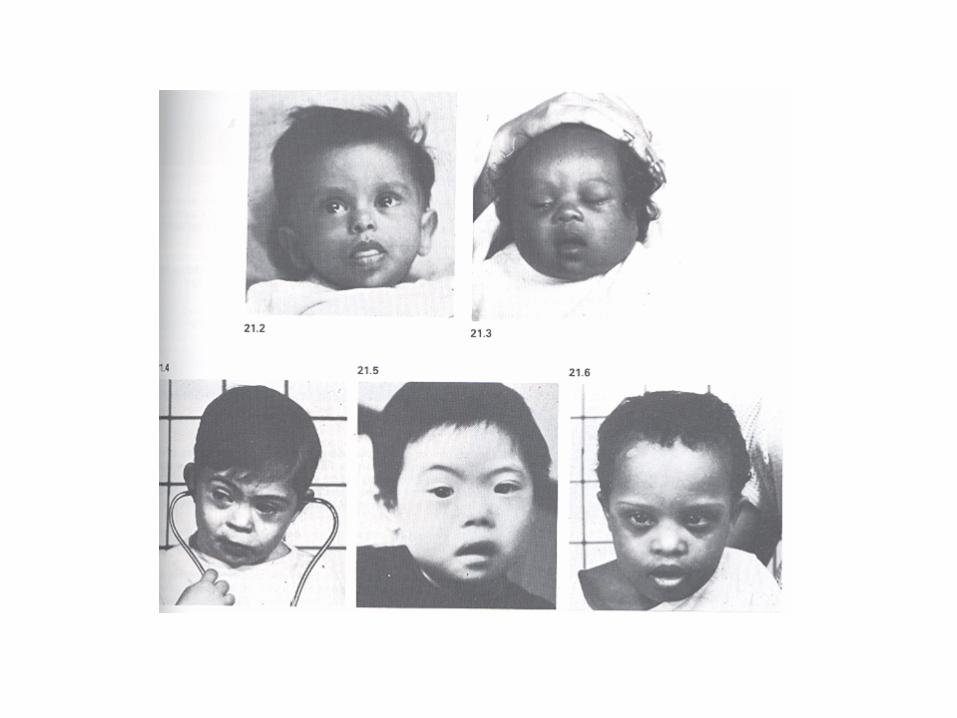

La SD si associa sovente a complicanze malformative che richiedono interventi chirurgici rilevanti nel corso dei primi anni di vita:
• il 50% presenta malformazioni cardiache, • il 30% stenosi duodenale, • l’1% atresia esofagea, • il 2% malformazioni anorettali. • La chirurgia oftalmica è richiesta nel 12% dei casi per problemi di cataratta.
Oltre alle malformazioni congenite descritte, il soggetto con SD ha la tendenza a sviluppare patologie secondarie per deficit nel sistema immunitario con particolare predisposizione ad infezioni batteriche; nell’1% poi dei casi compare leucemia acuta. Nel corso della vita il soggetto Down tende anche a sviluppare ipotiroidismo e diabete mellito.
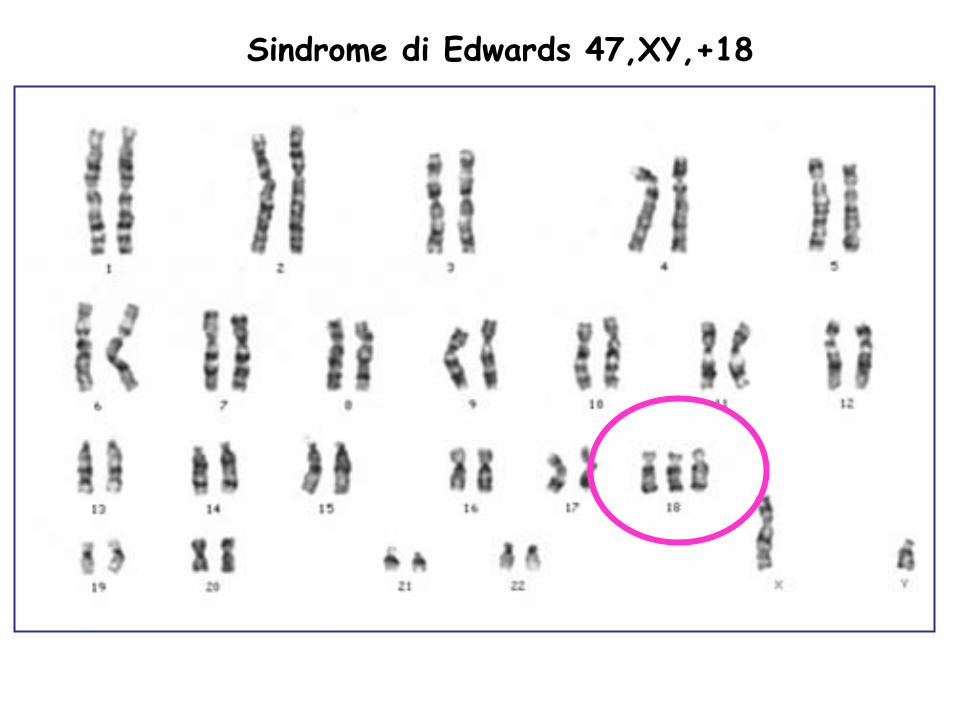
Sindrome di Edwards 47,XY,+18

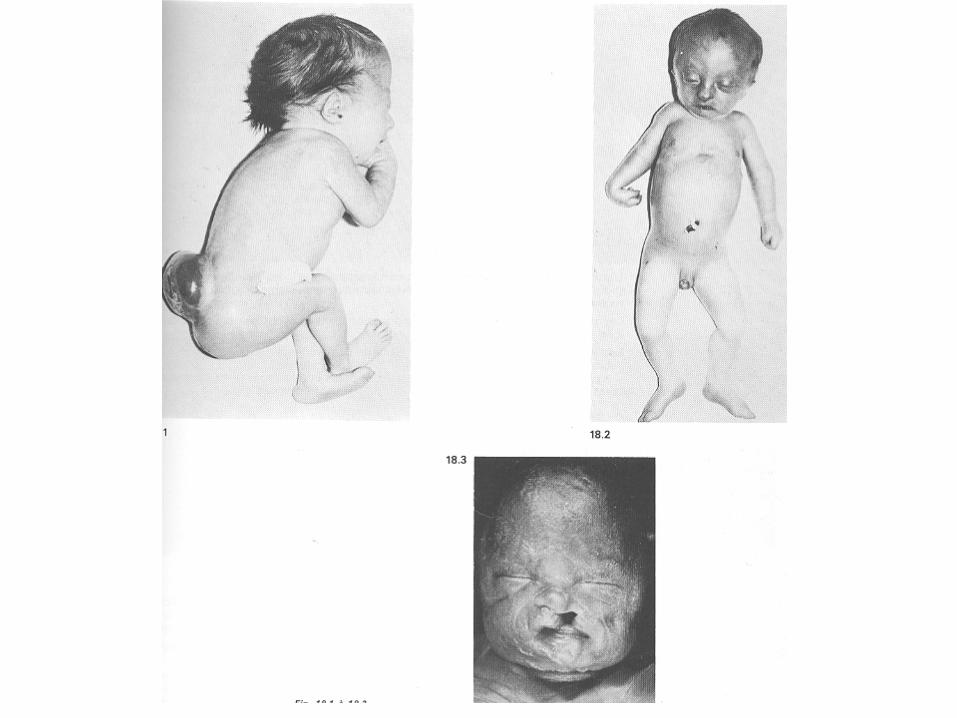
SINDROME DI EDWARDS
Frequenza alla nascita: 1:7000 nati vivi.
Circa il 50% degli affetti muore entro il 1°mese di vita e solo il 10% raggiunge il 1° anno.
C i rca i l 95% deg l i z i got i + 18 è abort i to spontaneamente.
Analogamente alla +21 è stata dimostrata una correlazione tra l’aneuploidia e l’età materna.

Caratteristiche fenotipiche della sindrome di Edwards: • orecchie faunesche • ritardo di sviluppo psico-motorio • micrognazia • difetti di chiusura del tubo neurale • accavallamento delle dita • piede torto • ipotonia importante • malformazioni cardiache e renali • morte precoce

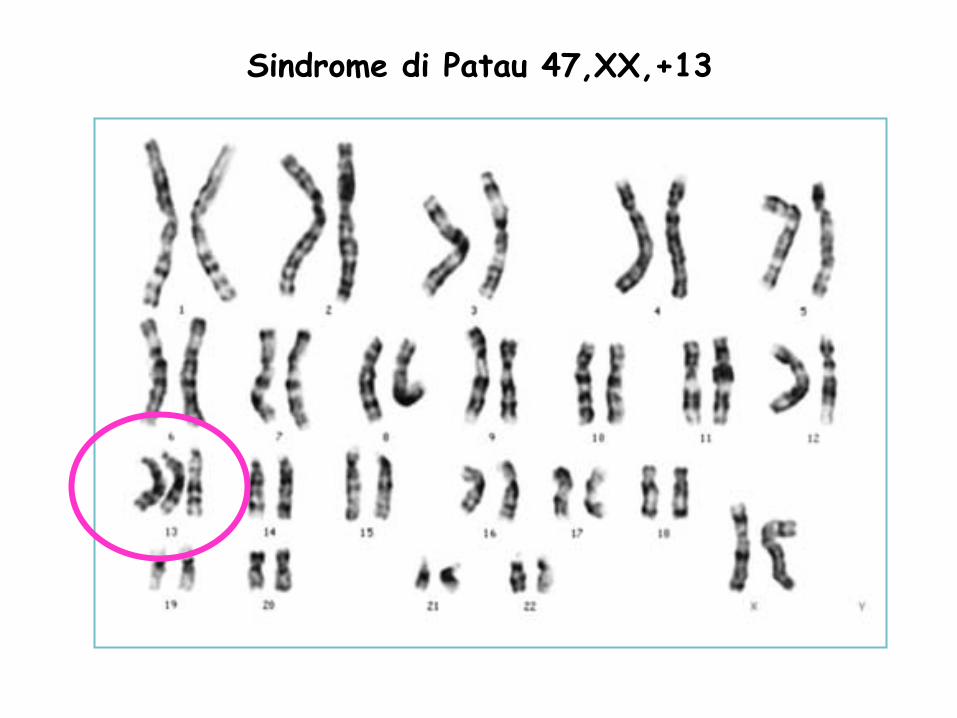
Sindrome di Patau 47,XX,+13

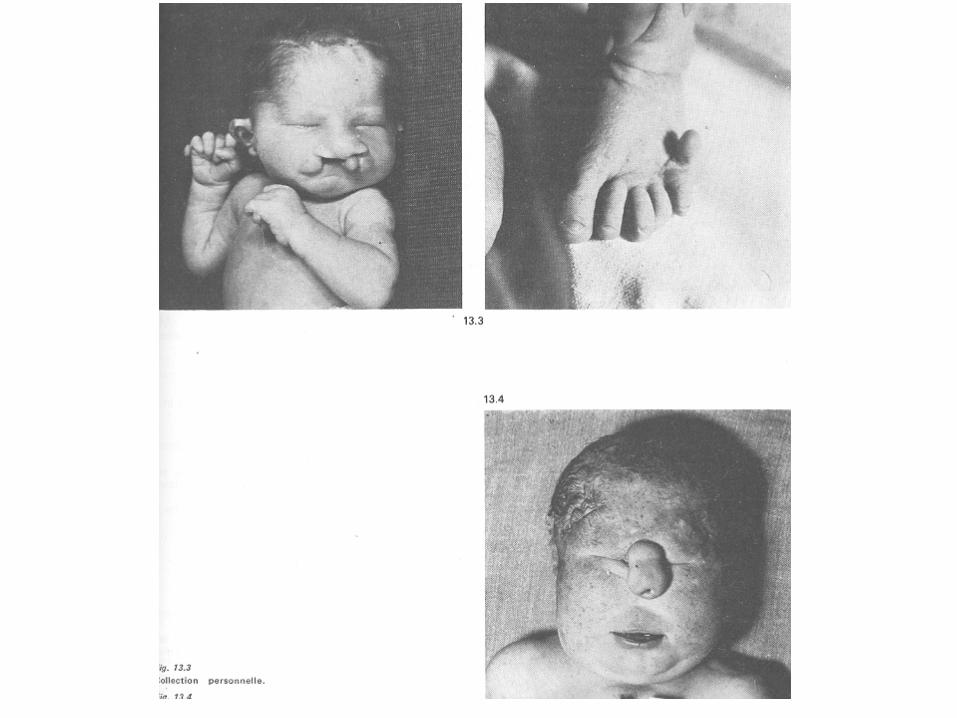
SINDROME DI PATAU
Alla nascita ha una frequenza di 1:10.000.
Le attese di vita sono analoghe a quelle osservate per la trisomia 18. Oltre il 95% delle trisomia 13 è abortito spontaneamente. Il 90% ha trisomia libera; negli altri casi la trisomia è trasmessa da un genitore portatore di una traslocazione robertsoniana coinvolgente il cromosoma 13.

Caratteristiche fenotipiche della sindrome di Patau : • labio-palotoschisi • microftalmia • esadattilia • ciclopia • ritardo psicomotorio • cardiopatia • encefalopatia • morte precoce
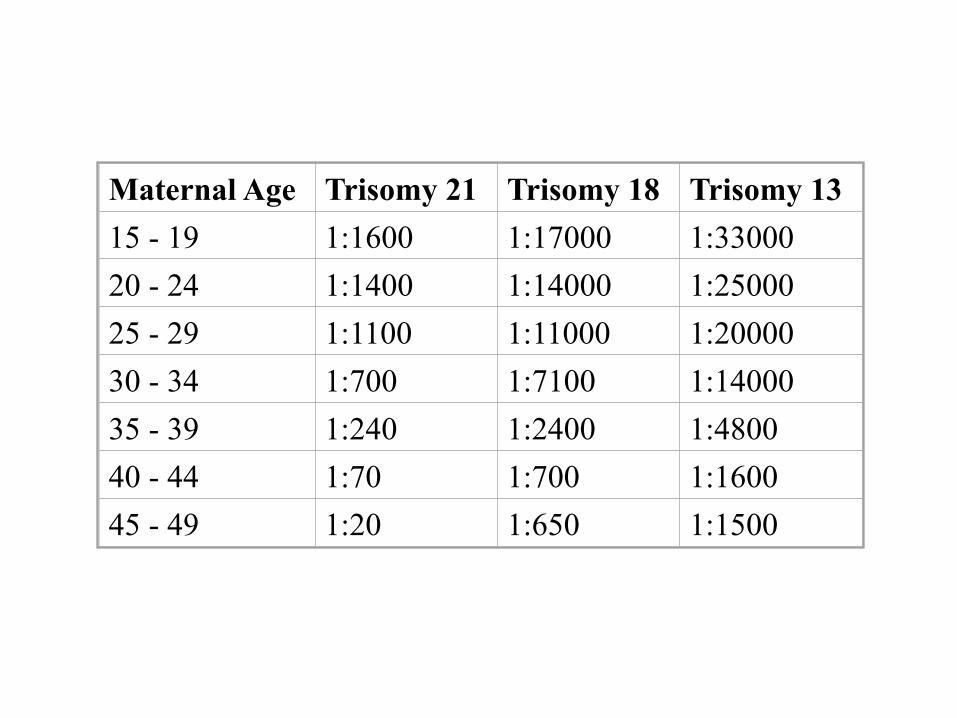
Maternal Age Trisomy 21 Trisomy 18 Trisomy 13 15 - 19 1:1600 1:17000 1:33000 20 - 24 1:1400 1:14000 1:25000 25 - 29 1:1100 1:11000 1:20000 30 - 34 1:700 1:7100 1:14000 35 - 39 1:240 1:2400 1:4800 40 - 44 1:70 1:700 1:1600 45 - 49 1:20 1:650 1:1500

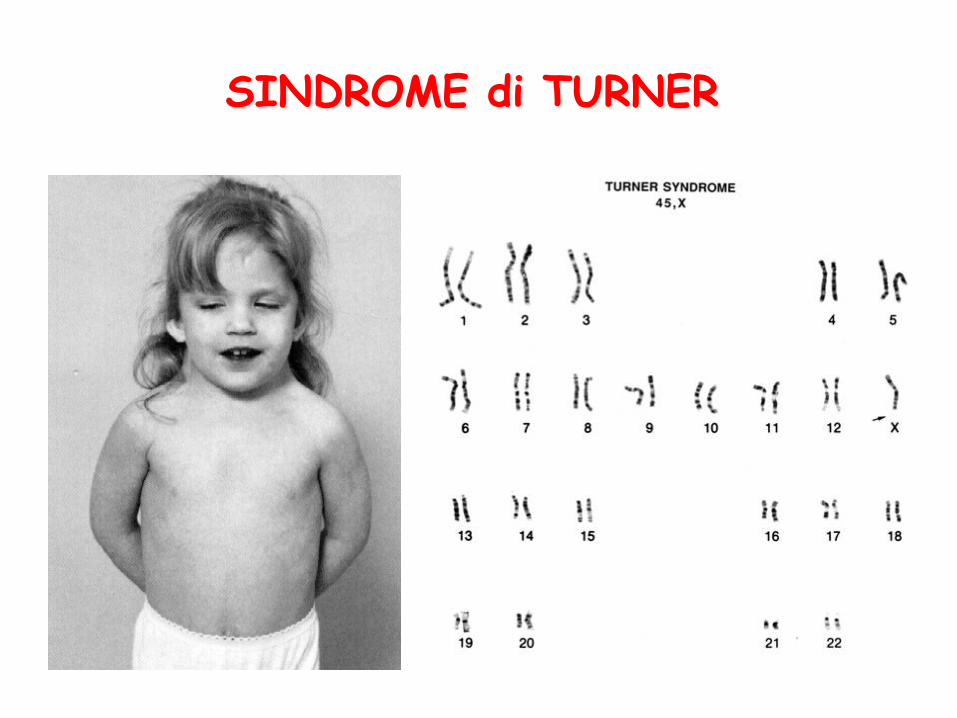
SINDROME di TURNER
Cariotipo 45,X (fenotipicamente femmine) Frequenza alla nascita: 1:5000 – 1:10.000 femmine. Circa il 99% degli zigoti è abortito, la frequenza dei prodotti del concepimento 45,X è alta. La diagnosi può essere fatta alla nascita per la presenza di cute in esubero sulla nuca, o può essere posta solo più tardi per bassa statura (inferiore a 140 cm), amenorrea primaria, collo a “tenda” (pterigio). Presentano infantilismo dei caratteri sessuali secondari. L’intelligenza è normale.

• Tessuto ovarico costituito da connettivo, caratteri sessuali poco sviluppati. (La somministrazione di ormoni sessuali migliora lo sviluppo dei caratteri sessuali secondari) • Cariotipo più frequente: 45,X (spermatogenesi) (Presenza di cariotipo a mosaico o anomalie strutturali del cromosoma X) • Questa anomalia non correla con l’età parentale. Non c’è rischio di ricorrenza nei genitori delle affette.
SINDROME di TURNER

SINDOME DI KLINEFELTER
• Cariotipo 47,XXY (fenotipicamente maschi)
• Incidenza 1: 1.000 maschi nati vivi. • Presentano ipogonadismo caratterizzato da bassi livelli di testosterone, testicoli di piccole dimensioni, azospermia costante*. Può essere presente ginecomastia. La maggior parte dei soggetti affetti presenta un’intelligenza nella norma *La diagnosi di solito viene posta durante le indagini per infertilità • Nel 50% dei casi l’errore è meiotico materno, ed è legato all’età.

FEMMINA XXX Frequenza alla nascita: 1:1000. Il fenotipo è normale, può essere presente raramente sterilità e irregolarità del ciclo. Le pazienti fertili hanno figli normali.
MASCHIO XYY Frequenza 1:1000. Non è associato ad un quadro clinico riconoscibile. La fertilità è normale.

ANEUPLOIDIE DEI CROMOSOMI SESSUALI Aneuploidie più compatibili con la vita
(ad eccezione della nullisomia – assenza - del cromosoma X) Perché?
• Polisomie X “compensate” dal processo di inattivazione del cromosoma X • La maggior parte del cromosoma Y è geneticamente “inerte” (eterocromatina costitutiva)
Ricorda! • La nullisomia del cromosoma X non permette lo sviluppo dell’embrione, anche in presenza del cromosoma Y (l’X è ricco di geni fondamentali per lo sviluppo dell’embrione che non hanno alleli omologhi sull’Y)
• L’X contiene circa 2000 geni attivi, l’Y meno di 100

Compensazione del dosaggio • Nelle specie in cui la femmine ha due X e il
maschio ha un solo X, esistono dei meccanismi di compensazione del dosaggio che rendono equivalente l’espressione dei geni X-linked nei due sessi
• In Drosophila il maschio incrementa l’attività del suo unico X
• Nei mammiferi la femmina inattiva uno dei due X











![Poliabortività 2012 [modalità compatibilità] ... · Anomalie cromosomiche 2,3 55% degli aborti sporadici 46% degli aborti ricorrenti All’ ↑ n° aborti in pz < 36aa la probabilità](https://static.fdocumenti.com/doc/165x107/5c18318409d3f2fa588c6e2e/poliabortivita-2012-modalita-compatibilita-anomalie-cromosomiche-23.jpg)





