
6.4 Processi di polimerizzazione - Treccani, il portale ... · cioè il tempo necessario a...
20
369 VOLUME V / STRUMENTI 6.4.1 Reazioni di polimerizzazione I polimeri sono macromolecole di origine naturale o sintetica che presentano diverse caratteristiche chimico-fisiche tali da consentirne l’applicazione in svariati settori delle attività umane. La produzione mondiale di polimeri è dell’ordine del miliar- do di tonnellate l’anno e coinvolge ormai tutti i settori indu- striali: dai trasporti ai prodotti alimentari, dall’edilizia ai mate- riali per l’elettronica, dal tessile al farmaceutico. I polimeri sono ottenuti a partire da molecole relativamente semplici, dette monomeri, che si assemblano tra loro con diver- si meccanismi e geometrie, costituendo macromolecole che possono inglobare da alcune decine (in questo caso si parla di oligomeri) fino a diverse migliaia di unità monomeriche. In generale questo processo di assemblaggio si svolge attraverso due possibili meccanismi chimici: quello di crescita a stadi e quello di crescita a catena. Alla luce delle reazioni chimiche che caratterizzano il processo di polimerizzazione è possibile procedere a una semplice classificazione dei polimeri sintetici. I più diffusi sono i polimeri termoplastici, costituiti da macromolecole lineari o ramificate, che presentano una temperatura di fusio- ne al di sopra della quale liquefanno in modo reversibile. In presenza di elevati gradi di reticolazione, e dunque di pesi molecolari molto più elevati, si ottengono i termoset che all’aumentare della temperatura si decompongono senza fon- dere. Infine gli elastomeri sono ancora macromolecole linea- ri o ramificate ma contengono al loro interno dei doppi lega- mi che vengono fatti reagire in un secondo tempo con l’ag- giunta di un opportuno agente reticolante ad alta temperatura, per ottenere un materiale reticolato con caratteristiche tipi- che delle gomme. Meccanismo di crescita a stadi Questo meccanismo di polimerizzazione è caratterizzato dal fatto che tutte le molecole presenti nell’ambiente di rea- zione (monomeri, oligomeri e polimeri) conservano la mede- sima probabilità di reagire durante l’intero processo. Questo è il caso dei poliesteri, che vengono sintetizzati attraverso la rea- zione di esterificazione a partire da monomeri che presentano due gruppi acidi (A) o due alcolici (B). Per esempio partendo da acido tereftalico e dietanolo si ottiene il polietilentereftala- to. In generale indicando con AA e BB i due monomeri, dove A e B rappresentano i gruppi acidi e alcolici, si ottiene la seguente stechiometria di reazione: [1] In modo analogo si possono rappresentare altre reazioni che portano alla sintesi di altri tipi di macromolecole, come nel caso delle poliammidi in cui i gruppi reagenti sono un’am- mina e un acido carbossilico, o i poliuretani, ottenuti per esem- pio a partire da un diolo e da un diisocianato. Lo stesso meccanismo di polimerizzazione si applica a monomeri che contengono entrambi i gruppi funzionali, come schematizzato dalla seguente stechiometria di reazione: [2] Questo è per esempio il caso della reazione tra amminoa- cidi in cui il legame peptidico, tipico della struttura delle pro- teine, viene formato a partire da un gruppo amminico e uno carbossilico: [3] Alcune di queste reazioni sono riportate in tab. 1, dove è possibile osservare che spesso l’addizione di un’unità mono- merica coinvolge l’eliminazione di una molecola di basso peso molecolare, come per esempio acqua o metanolo. Questa è una caratteristica importante di tale tipo di polimerizzazioni, che per essere portate a completamento richiedono l’allontana- mento dal luogo di reazione di queste specie a basso peso mole- colare. Come appare chiaramente dalla stechiometria di reazione [1], le unità di monomero si alternano lungo la catena. Pertanto per ottenere conversione completa e dunque macromolecole a elevato peso molecolare è necessario operare con esatta equi- valenza dei gruppi funzionali. Dal punto di vista cinetico la caratteristica principale del meccanismo di crescita a stadi è che le catene polimeriche rimangono attive, e dunque continuano a crescere, durante l’in- tero processo di polimerizzazione, cioè per tutto il tempo neces- sario a consumare completamente le specie monomeriche. In realtà poiché i gruppi reattivi rimangono inalterati nelle catene | | ➤ H N R COHN R' COOH + H 2 68 68 68 68 2 O H N R COOH + H N R' COOH 2 2 68 68 68 68 ➤ || n n A A B A B A 68 68 68 68 68 6 B ( ) ( ) − ➤ || 2 8 B B B 68 6 ➤ || 8 68 68 68 68 68 A A B B A A ( ) − n 1 n n A A B B 68 68 ( ) + ( ) ➤ || 6.4 Processi di polimerizzazione
-
Upload
truongxuyen -
Category
Documents
-
view
224 -
download
0
Transcript of 6.4 Processi di polimerizzazione - Treccani, il portale ... · cioè il tempo necessario a...

369VOLUME V / STRUMENTI
6.4.1 Reazioni di polimerizzazione
I polimeri sono macromolecole di origine naturale o sinteticache presentano diverse caratteristiche chimico-fisiche tali daconsentirne l’applicazione in svariati settori delle attività umane.La produzione mondiale di polimeri è dell’ordine del miliar-do di tonnellate l’anno e coinvolge ormai tutti i settori indu-striali: dai trasporti ai prodotti alimentari, dall’edilizia ai mate-riali per l’elettronica, dal tessile al farmaceutico.
I polimeri sono ottenuti a partire da molecole relativamentesemplici, dette monomeri, che si assemblano tra loro con diver-si meccanismi e geometrie, costituendo macromolecole chepossono inglobare da alcune decine (in questo caso si parla dioligomeri) fino a diverse migliaia di unità monomeriche. Ingenerale questo processo di assemblaggio si svolge attraversodue possibili meccanismi chimici: quello di crescita a stadi equello di crescita a catena.
Alla luce delle reazioni chimiche che caratterizzano ilprocesso di polimerizzazione è possibile procedere a unasemplice classificazione dei polimeri sintetici. I più diffusisono i polimeri termoplastici, costituiti da macromolecolelineari o ramificate, che presentano una temperatura di fusio-ne al di sopra della quale liquefanno in modo reversibile. Inpresenza di elevati gradi di reticolazione, e dunque di pesimolecolari molto più elevati, si ottengono i termoset cheall’aumentare della temperatura si decompongono senza fon-dere. Infine gli elastomeri sono ancora macromolecole linea-ri o ramificate ma contengono al loro interno dei doppi lega-mi che vengono fatti reagire in un secondo tempo con l’ag-giunta di un opportuno agente reticolante ad alta temperatura,per ottenere un materiale reticolato con caratteristiche tipi-che delle gomme.
Meccanismo di crescita a stadiQuesto meccanismo di polimerizzazione è caratterizzato
dal fatto che tutte le molecole presenti nell’ambiente di rea-zione (monomeri, oligomeri e polimeri) conservano la mede-sima probabilità di reagire durante l’intero processo. Questo èil caso dei poliesteri, che vengono sintetizzati attraverso la rea-zione di esterificazione a partire da monomeri che presentanodue gruppi acidi (A) o due alcolici (B). Per esempio partendoda acido tereftalico e dietanolo si ottiene il polietilentereftala-to. In generale indicando con A�A e B�B i due monomeri,
dove A e B rappresentano i gruppi acidi e alcolici, si ottiene laseguente stechiometria di reazione:
[1]
In modo analogo si possono rappresentare altre reazioniche portano alla sintesi di altri tipi di macromolecole, comenel caso delle poliammidi in cui i gruppi reagenti sono un’am-mina e un acido carbossilico, o i poliuretani, ottenuti per esem-pio a partire da un diolo e da un diisocianato.
Lo stesso meccanismo di polimerizzazione si applica amonomeri che contengono entrambi i gruppi funzionali, comeschematizzato dalla seguente stechiometria di reazione:
[2]
Questo è per esempio il caso della reazione tra amminoa-cidi in cui il legame peptidico, tipico della struttura delle pro-teine, viene formato a partire da un gruppo amminico e unocarbossilico:
[3]
Alcune di queste reazioni sono riportate in tab. 1, dove èpossibile osservare che spesso l’addizione di un’unità mono-merica coinvolge l’eliminazione di una molecola di basso pesomolecolare, come per esempio acqua o metanolo. Questa è unacaratteristica importante di tale tipo di polimerizzazioni, cheper essere portate a completamento richiedono l’allontana-mento dal luogo di reazione di queste specie a basso peso mole-colare.
Come appare chiaramente dalla stechiometria di reazione[1], le unità di monomero si alternano lungo la catena. Pertantoper ottenere conversione completa e dunque macromolecole aelevato peso molecolare è necessario operare con esatta equi-valenza dei gruppi funzionali.
Dal punto di vista cinetico la caratteristica principale delmeccanismo di crescita a stadi è che le catene polimericherimangono attive, e dunque continuano a crescere, durante l’in-tero processo di polimerizzazione, cioè per tutto il tempo neces-sario a consumare completamente le specie monomeriche. Inrealtà poiché i gruppi reattivi rimangono inalterati nelle catene
||||||||��➤ H N R COHN R' COOH + H26888 6888 6888 6888
22O
H N R COOH + H N R' COOH 2 26888 6888 6888 6888 ��➤||||||||||
nn
A A B A B A6888 6888 6888 6888 6888 6B( ) ( ) −��➤||||||||
2
8888B
B B6888 6 ��➤|||||||| 8888 6888 6888 6888 6888 6888A A B B A A( ) −n 1
n nA A B B6888 6888( )+ ( ) ��➤||||||||
6.4
Processi di polimerizzazione

polimeriche finali, queste possono riprendere a crescere in qua-lunque momento se poste in contatto con i monomeri. Per que-sto motivo tali polimeri sono detti viventi, e questa caratteri-stica viene sfruttata in numerose loro applicazioni.
Nel caso di monomeri contenenti due gruppi reattivi siottengono catene lineari, come evidenziato dalla relazione [1].Nel caso invece di monomeri contenenti tre o più gruppi reat-tivi, si ottengono catene non lineari, che possono esibire strut-ture anche molto diverse. Una situazione tipica è la formazio-ne di strutture a elevatissimo peso molecolare, in cui le catenesono fortemente interconnesse e formano una fase macrosco-pica che ha caratteristiche chimico-fisiche completamentediverse da quelle della miscela dei monomeri di partenza o deicorrispondenti polimeri lineari. Tali macromolecole sono spes-so chiamate geli e possono essere separate dal resto della misce-la reagente in quanto risultano insolubili in qualunque solven-te. Un’altra struttura peculiare è quella di copolimeri sintetiz-zati a partire da monomeri che contengono due o più funzionalitàdi tipo A e monomeri che contengono due o più funzionalitàdi tipo B, che reagiscono tra loro ma non con se stesse e dovealmeno uno dei due monomeri possiede come minimo tre ditali funzionalità. In questo caso si ottengono polimeri iperre-ticolati che hanno strutture relativamente compatte ma con ele-vatissimi pesi molecolari.
Un esempio di questo tipo è il policarbosilano ottenuto persintesi di specie monomeriche, in cui i gruppi A e B di cui soprarappresentano un gruppo CH�CH2 e un gruppo Si�H. Que-ste strutture risultano particolarmente efficaci per vari tipi diapplicazioni nel settore dei prodotti per rivestimenti superfi-ciali, in quanto rispetto a polimeri lineari di analoga composi-zione chimica presentano, a parità di peso molecolare, unaviscosità assai inferiore.
Meccanismo di crescita a catenaQuesto meccanismo di formazione dei polimeri è caratte-
rizzato da tre processi in sequenza: il primo è l’iniziazione incui viene prodotto un centro attivo, che può essere un radica-le, un catione o un anione; su tale centro avviene la reazione
di addizione della specie monomerica che comporta anche laformazione di un analogo centro attivo sull’unità addizionata(ciò consente l’addizione successiva di varie unità monomeri-che e dunque la formazione della catena polimerica); infine sihanno le reazioni che portano all’interruzione della propaga-zione della catena, e quindi alla produzione della catena poli-merica definitiva o morta, cioè non più in grado di addiziona-re unità monomeriche. Queste reazioni vengono dette termi-nazioni se comportano la scomparsa del centro attivo otrasferimenti di catena se lo stesso viene semplicemente tra-sferito a un’altra molecola, consentendo cosi la propagazionedi una nuova catena.
Dal punto di vista cinetico la caratteristica fondamentaledel meccanismo di crescita a catena è che il tempo di vita dellacatena in crescita, cioè il tempo che intercorre tra l’attivazio-ne di una catena e la sua terminazione, è molto breve rispettoalla durata del processo di polimerizzazione. Pertanto, a diffe-renza del meccanismo di crescita a stadi, nuovi centri attividevono essere continuamente prodotti durante il processo, inmodo da rimpiazzare quelli soppressi dalle reazioni di termi-nazione e consentire al processo stesso di completarsi.
Come accennato, a seconda della natura del centro attivoche consente la propagazione della catena polimerica, si pos-sono avere polimerizzazioni radicaliche, cationiche o anioni-che. Anche alcune polimerizzazioni catalitiche, come quelledelle olefine su catalizzatori Ziegler-Natta, seguono un mec-canismo di reazione analogo, detto anche per coordinazione.In tab. 2 sono elencati alcuni polimeri che vengono prodottisecondo questi diversi meccanismi a catena. Nel seguito verràapprofondita la trattazione cinetica della polimerizzazione radi-calica, che è certamente la più diffusa.
6.4.2 Cinetica della polimerizzazione a stadi
Nella polimerizzazione a stadi il tempo di vita delle catene poli-meriche è confrontabile con il tempo caratteristico del processo,
ASPETTI PROCESSISTICI
370 ENCICLOPEDIA DEGLI IDROCARBURI
tab. 1. Polimeri prodotti attraverso la polimerizzazione a stadi
Poliestere
Poliammide
Poliuretano
Poliestere
HOOCn COOH OH HO C O HCO
O
� �n HO (2 n�1)H2O(CH2)2 (CH2)2
O n
HOOCn COOH �n H2N NH2(CH2)4 (CH2)6 HO C
H
CN
O
�(2n�1)H2O(CH2)6(CH2)4
O
H
HN
n
(n�1) HO OH �n OCN(CH2)4 (CH2)6 HO
H
OCN OH(CH2)6(CH2)4 (CH2)4
O
H
NCO
O
NCO
n
n HO COOH n HO HCO � (n�1)H2O
O n
cioè il tempo necessario a consumare completamente le spe-cie monomeriche. Pertanto, a differenza della polimerizzazio-ne radicalica, in questo caso il peso molecolare di tutte le cate-ne cresce in modo mediamente uniforme durante l’intera dura-ta del processo. Ciò implica che elevati pesi molecolari vengonoraggiunti solo per conversioni molto elevate, e pertanto per lamaggior parte del processo la viscosità rimane moderata e con-seguentemente i processi di scambio termico risultano relati-vamente agevoli.
Considerando due monomeri bifunzionali, come per esem-pio un diolo e un diacido, la reazione avviene secondo la ste-chiometria di reazione [1]. Nella trattazione cinetica di questiprocessi viene usualmente assunto che la reattività dei gruppifunzionali sia indipendente dalla lunghezza della catena a cuisono legati. Quando si verifica il caso, assai frequente nelleapplicazioni, in cui la concentrazione molare iniziale dei duemonomeri è uguale, la conversione dei gruppi funzionaliXA�(NA0�NA)�NA0, dove NA e NA0 rappresentano il numero dimoli del diacido, al generico tempo t e al tempo iniziale t�0rispettivamente, è data da
[4]
dove k è la costante cinetica del secondo ordine della reazionedi esterificazione [1] e A0 è la concentrazione iniziale del dia-cido.
In questi sistemi la lunghezza di catena media numerale,cioè il numero di unità mediamente presenti in una catena, nN,è data semplicemente dal rapporto tra il numero di moli di Aall’inizio e al generico tempo t del processo, che in termini diconversione porta a
[5]
da cui emerge come valori elevati del peso molecolare siano otte-nibili solo per conversioni molto elevate. Inoltre, nel caso in cuisia presente una deviazione anche piccola dalla composizionestechiometrica dei due monomeri, il peso molecolare massimoammissibile diminuisce significativamente. Conseguentementeper ottenere polimeri a elevato peso molecolare vengono nor-malmente preferite le tecniche di polimerizzazione a catena.
Per stimare la distribuzione delle lunghezze delle cate-ne polimeriche, è necessario valutare la probabilità che unacatena sia costituita da r unità monomeriche. Questa puòessere ottenuta considerando che tale catena deve essere co-stituita da (r�1) gruppi funzionali che hanno reagito e dauno che non ha reagito. Poiché la probabilità che un gruppo
funzionale selezionato a caso nella miscela reagente abbiareagito è data dalla conversione, XA, è possibile valutare lafrazione di catene N(r) che hanno una lunghezza assegnatar come segue:
[6]
Da questa è possibile valutare la frazione di unità mono-meriche contenute in catene di lunghezza r
[7]
il cui valore medio è dato da:
[8]
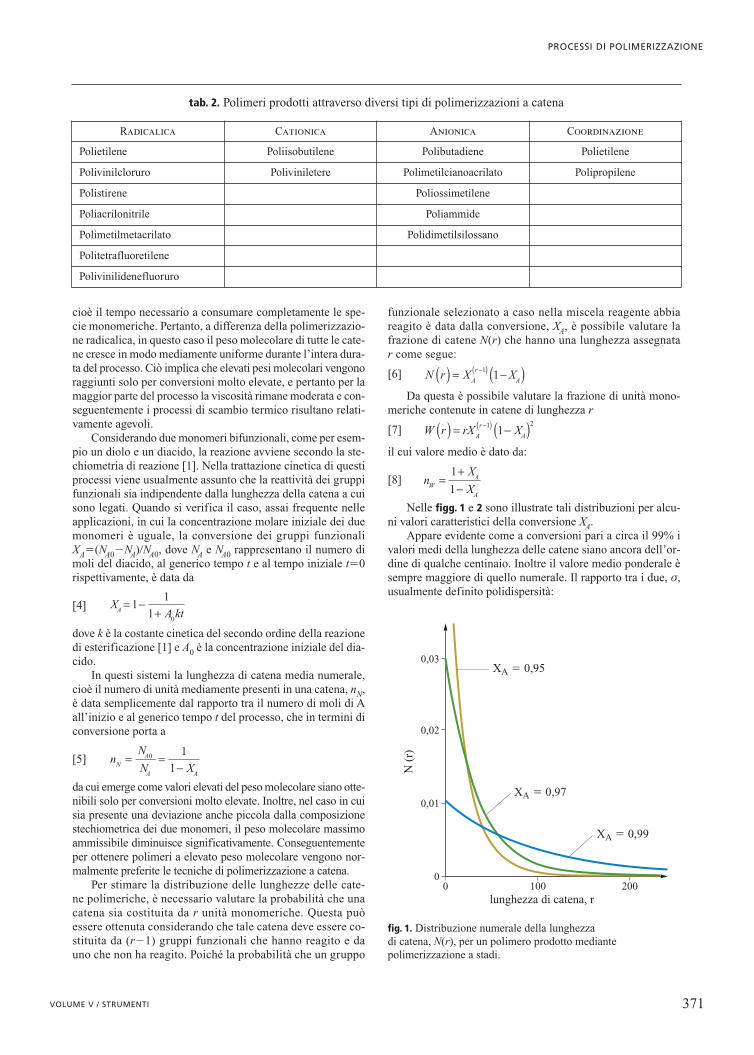
Nelle figg. 1 e 2 sono illustrate tali distribuzioni per alcu-ni valori caratteristici della conversione XA.
Appare evidente come a conversioni pari a circa il 99% ivalori medi della lunghezza delle catene siano ancora dell’or-dine di qualche centinaio. Inoltre il valore medio ponderale èsempre maggiore di quello numerale. Il rapporto tra i due, s,usualmente definito polidispersità:
nXXW
A
A
=+−
1
1
W r rX XAr
A( ) = −( )−( )1 2
1
N r X XAr
A( ) = −( )−( )11
nNN XN
A
A A
= =−
01
1
XA ktA = −
+1
1
10
PROCESSI DI POLIMERIZZAZIONE
371VOLUME V / STRUMENTI
XA � 0,95
XA � 0,97
XA � 0,99
N (
r)
0,01
0
0,02
0,03
lunghezza di catena, r1000 200
fig. 1. Distribuzione numerale della lunghezza di catena, N(r), per un polimero prodotto mediantepolimerizzazione a stadi.
tab. 2. Polimeri prodotti attraverso diversi tipi di polimerizzazioni a catena
Radicalica Cationica Anionica Coordinazione
Polietilene Poliisobutilene Polibutadiene Polietilene
Polivinilcloruro Poliviniletere Polimetilcianoacrilato Polipropilene
Polistirene Poliossimetilene
Poliacrilonitrile Poliammide
Polimetilmetacrilato Polidimetilsilossano
Politetrafluoretilene
Polivinilidenefluoruro

[9]
fornisce una stima dell’ampiezza della distribuzione. All’ini-zio del processo, XA�0 e s�1, essendo tutte le catene (mono-meri) di lunghezza uguale. All’aumentare della conversione lapolidispersità aumenta, mantenendo però valori assai conte-nuti. A conversione completa (XA�1) si ottiene infatti s�2 che,confrontato per esempio con i valori tipici dei processi di poli-merizzazione a catena, risulta assai contenuto.
È possibile concludere che i processi di polimerizzazionea stadi, come tutti i processi viventi, sono caratterizzati da gran-de uniformità delle catene polimeriche. Ciò vale in particola-re per la distribuzione della lunghezza delle catene polimeri-che che risulta in generale stretta, anche se caratterizzata davalori medi relativamente bassi. Questi diminuiscono ulterior-mente in presenza di scostamenti dalla composizione stechio-metrica dei due monomeri o di impurezze.
6.4.3 Polimerizzazione radicalica a catena
IniziazioneLa reazione di generazione di radicali più frequentemente
adottata è quella di decomposizione termica di una specie chi-mica relativamente instabile, quale per esempio un perossido.La scelta dell’iniziatore viene generalmente effettuata sullabase della temperatura e della durata del processo, in modo dagarantire la necessaria produzione di radicali fino al comple-to consumo delle specie monomeriche.
La stechiometria di decomposizione tipica di un perossi-do è la seguente:
[10]
Tuttavia, dei radicali prodotti da questa o da simili reazio-ni di decomposizione, solo una frazione dà luogo a una cate-na in grado di propagare. Ciò viene indicato da una grandez-za detta efficienza dell’iniziatore ( f ) che assume valori com-presi tra 0,2 e 1. Il maggiore responsabile di questo fenomenoè la reazione di ricombinazione dei due radicali appena for-mati. Questa reazione è favorita dal cosiddetto ‘effetto gabbia’,
dovuto alla gabbia formata dalle molecole di solvente e mono-mero che circondano le molecole di iniziatore al momento dellaloro decomposizione. I due radicali formati devono infatti riu-scire a diffondere fuori da tale gabbia prima che si verifichi lareazione di ricombinazione. Si avranno dunque in generalevalori di efficienza più bassi all’aumentare della viscosità delsistema reagente, e dunque al procedere della polimerizzazio-ne. È da notare che la reazione di ricombinazione non portasempre alla ricostituzione della molecola originaria di inizia-tore, che sarebbe dunque nuovamente in grado di produrre radi-cali. Questo è per esempio il caso dell’azoisobutirronitrile(AIBN), la cui decomposizione avviene attraverso l’elimina-zione di una molecola di azoto.
Una situazione particolare si presenta nel caso delle poli-merizzazioni che devono essere condotte a basse temperature,per le quali è difficile trovare iniziatori in grado di produrre iradicali sufficienti per sostenere lo svolgimento della reazio-ne di polimerizzazione. In questo caso è possibile accelerarela reazione di decomposizione dell’iniziatore per via fotochi-mica, utilizzando radiazioni visibili o ultraviolette. Alternati-vamente è possibile catalizzare la reazione con un agente ridu-cente, per esempio un sale di Fe2�, dando luogo a una reazio-ne di ossidoriduzione che porta alla formazione di una specieradicalica e che diventa dominante rispetto alla decomposi-zione non catalizzata [10]
[11]
Radiazioni i cui fotoni sono sufficientemente energetici,come UV o raggi gamma, possono essere utilizzate per iniziarele reazioni di polimerizzazione radicalica, anche in assenza diiniziatore. Questo metodo, che ha il vantaggio di essere del tuttoindipendente dalla temperatura, viene utilizzato in particolarenei processi di post-trattamento per reticolazione dei polimeri.
A temperature sufficientemente elevate può verificarsi chele stesse molecole di monomero diano luogo a reazioni di decom-posizione con formazione di radicali. Un esempio tipico è lo sti-rene che, attraverso un meccanismo sufficientemente comples-so, è in grado di dare luogo a una reazione di produzione di radi-cali che segue una cinetica del terzo ordine rispetto allaconcentrazione di monomero. Questa reazione viene utilizzatanei processi di polimerizzazione in massa ad alta temperaturadello stirene, impiegati per la produzione di specie oligomeriche.
In generale la decomposizione del monomero con forma-zione di radicali è in realtà una reazione indesiderata. Essa puòinfatti verificarsi durante le fasi di stoccaggio o di trasportodei monomeri portando, in tempi generalmente lunghi, allaloro polimerizzazione. Per evitare ciò vengono introdotti alcu-ni inibitori di polimerizzazione. Questi sono specie chimiche,come per esempio gli idrochinoni, in grado di reagire rapida-mente con i radicali eventualmente formati producendo spe-cie ancora radicaliche ma stabili e dunque non in grado di ini-ziare una catena di reazioni radicaliche.
PropagazioneLe specie chimiche più utilizzate nei processi di polime-
rizzazione sono i monomeri vinilici, costituiti da una mole-cola di etilene in cui uno solo dei due atomi di carbonio hauno o due sostituenti (CH2�CHR o CH2�CRR') così danon inibire stericamente in modo eccessivo la reazione di aper-tura del doppio legame. Nella reazione di propagazione il radi-cale sulla catena in crescita apre il doppio legame del mono-mero, addizionandolo alla catena e trasferendogli l’attivitàradicalica:
Fe RO OR' Fe RO R'O2 3+ ++ + +6888 �� � �➤||||||||
RO OR' RO R'O6888
kd���� � �➤|||||||| +
σ = = +nn
XW
NA1
ASPETTI PROCESSISTICI
372 ENCICLOPEDIA DEGLI IDROCARBURI
W(r
). 102
1,0
0
2,0
lunghezza di catena, r1000 200 300
XA � 0,95
XA � 0,97
XA � 0,99
fig. 2. Distribuzione ponderale della lunghezza di catena, W(r), per un polimero prodotto mediantepolimerizzazione a stadi.

[12]
La reazione di propagazione segue in generale una cine-tica di tipo Arrhenius. Essendo infatti la specie monomeri-ca in grado di diffondere assai più rapidamente delle gran-di macromolecole radicaliche, i processi diffusivi non risul-tano limitare la cinetica di queste reazioni. Ciò viene tuttaviameno quando la temperatura di reazione scende sotto la tem-peratura di transizione vetrosa, e il sistema reagente passadallo stato di liquido viscoso a quello di solido. In questecondizioni la diffusione del monomero è fortemente inibi-ta e la reazione di propagazione, e con essa l’intero processodi polimerizzazione, si blocca. Questo fenomeno è respon-sabile dei numerosi casi riscontrati nelle applicazioni di que-ste reazioni, nei quali non è possibile raggiungere la con-versione completa.
Un altro aspetto potenzialmente rilevante in questo conte-sto è la reazione di depropagazione. Il valore di temperatura acui la velocità di questa reazione risulta uguale a quella dellareazione di propagazione, e dunque si raggiungono condizio-ni di equilibrio, è detto ceiling temperature. Utilizzando l’u-suale condizione di equilibrio termodinamico e indicando conDG(T) la variazione di energia libera di Gibbs legata alla rea-zione di propagazione, si ottiene:
[13]
dove M è la concentrazione del monomero. Dalla [13] appareevidente che tale temperatura di equilibrio dipende dalla con-centrazione di monomero presente nel sistema reagente. Èopportuno notare che la maggior parte dei processi di polime-rizzazione è condotta a temperature tali che la reazione di depro-pagazione risulta trascurabile fino a conversione praticamen-te completa del monomero.
TerminazioniLe reazioni responsabili della soppressione di un radica-
le, trascurando la possibile presenza di inibitori o impurezze,sono le terminazioni bimolecolari tra due catene radicalichein crescita. La reazione di questo tipo più frequente è la com-binazione dei due radicali con formazione di un’unica catenamorta:
[14]
Una reazione alternativa è la terminazione per dispropor-zionamento attraverso la quale una delle due catene radicali-che strappa un atomo di idrogeno dall’altra, inducendo su diessa la formazione di un doppio legame terminale:
[15]
Si noti che in questo caso le due catene terminano mante-nendo inalterata la loro individualità, e in particolare la lorolunghezza, a differenza di quanto accade nel caso della termi-nazione per combinazione.
Dal punto di vista cinetico la terminazione per combina-zione presenta la peculiarità di essere controllata dai fenome-ni diffusivi, fatto usualmente indicato come effetto Tromm-sdorff. Si tratta di una reazione caratterizzata da un atto reat-tivo, la combinazione dei due radicali, estremamente veloce.Questo deve essere però preceduto da un processo diffusivoassai complesso che deve portare alla sovrapposizione di duespecifici punti (i radicali) di due macromolecole aventi unaconformazione di tipo a gomitolo. I movimenti relativi di que-ste due molecole sono fortemente rallentati dalle numeroseinterazioni tra gli atomi delle due molecole reagenti e con quel-li delle altre macromolecole che le circondano. Il risultato del-l’effetto Trommsdorff è una costante cinetica che, al contrariodi quella classica di Arrehnius, presenta una forte dipendenzadalla viscosità del sistema reagente e una assai più modestadipendenza dalla temperatura, tipica dei processi di diffusio-ne materiale. Inoltre tale costante cinetica risulta essere pocosensibile alla natura chimica delle specie radicaliche in giocoma assai di più alle loro dimensioni.
Anche la terminazione per disproporzionamento risulta inalcuni casi controllata dall’effetto Trommsdorff, anche se piùraramente rispetto a quella per combinazione. Ciò a causa del-l’atto reattivo che, coinvolgendo la rottura di un legame C�H,risulta significativamente rallentato.
Trasferimento di catenaSi tratta di reazioni che arrestano la crescita della catena
radicalica, ma non sopprimono l’attività radicalica, trasferen-dola a un’altra specie chimica da cui comincia a propagare unanuova catena. Nel caso in cui il radicale non muti la propria atti-vità nel trasferimento, la velocità di consumo del monomero, edunque la durata del processo di polimerizzazione, non sonoalterate da questa reazione, che incide viceversa sulla lunghez-za delle catene polimeriche prodotte. In considerazione deglieffetti che le reazioni di trasferimento possono avere sulla strut-tura delle molecole prodotte è bene distinguere diversi casi.
Si parla di reazioni di trasferimento a piccole molecole quan-do il radicale viene trasferito a una molecola di solvente, dimonomero o di trasferitore di catena, che rappresenta una spe-cie appositamente aggiunta nell’ambiente di reazione per limi-tare la lunghezza delle macromolecole prodotte. Si noti che a
ktd
R RR R
n�1 m�1
R
ktd
R
n�1 m�1
ktc
R
m�n�1
R
ktc
R
m�1n�1
∆G T RT M( ) = ln
k p
RR
n
R
kp
n�1
PROCESSI DI POLIMERIZZAZIONE
373VOLUME V / STRUMENTI

ognuna di tali reazioni corrisponde spesso un diverso gruppoterminale sulla catena polimerica finale. La misura della distri-buzione di tali unità terminali è spesso un ottimo strumento peridentificare e quantificare la cinetica di queste reazioni.
La reazione di trasferimento di catena a monomero puòseguire la seguente stechiometria:
[16]
La specie radicalica formata porta, dopo l’addizione di unnumero opportuno di unità monomeriche, a una catena poli-merica morta caratterizzata da un doppio legame terminale,del tutto analoga a quella prodotta dalla reazione di termina-zione per disproporzionamento.
È opportuno osservare che le reazioni di propagazione e ditrasferimento a monomero coinvolgono gli stessi reagenti e nonsono pertanto separabili, ma avvengono sempre contemporanea-mente e proporzionalmente alle rispettive costanti cinetiche. Perogni data specie monomerica la reazione di trasferimento a mono-mero stabilisce dunque il peso molecolare massimo raggiungibi-le, nel caso in cui fosse possibile sopprimere ogni altra termi-nazione. Poiché la rottura del legame C�H richiesto dalla rea-zione di trasferimento [16] è certamente molto più difficiledell’apertura del doppio legame coinvolta nella reazione di pro-pagazione [12], tra le due reazioni esiste per i monomeri normal-mente utilizzati una differenza di almeno tre ordini di grandezza.
Le reazioni di trasferimento possono coinvolgere anche lemacromolecole, come nel caso della reazione di trasferimen-to a polimero:
[17]
In questo caso il radicale in crescita strappa un idrogenoda un punto qualsiasi lungo una catena morta, lasciandovi unradicale che può propagare creando una nuova catena. Poichéperò la nuova catena è unita alla precedente in un punto chenella reazione [17] è rappresentato da un carbonio terziario, siforma un ramo laterale.
Questa reazione presenta due importanti peculiarità rispet-to a tutte le reazioni finora considerate: la capacità di riattiva-re una catena morta e la formazione di catene non lineari o
ramificate. Ciò complica notevolmente la descrizione dellacinetica del processo e amplia enormemente la varietà di strut-ture realizzabili, rendendone anzi irrealistica una descrizionedettagliata in termini matematici.
Poiché la reazione di trasferimento può avvenire su unaqualsiasi delle unità monomeriche presenti lungo la catena poli-merica morta, la corrispondente velocità di reazione risultaproporzionale alla concentrazione di massa e non a quella mola-re. Ciò spiega anche come mai la reazione di trasferimento apolimero, pur coinvolgendo due macromolecole, non sia con-trollata dall’effetto Trommsdorff. Pur essendo infatti l’atto reat-tivo molto simile a quello della reazione di disproporziona-mento, il processo diffusivo risulta assai facilitato dal fatto chela catena morta può reagire in qualsiasi suo punto.
La reazione di trasferimento a polimero può anche essereintramolecolare, cioè il radicale posto all’estremità di una cate-na può strappare un atomo di idrogeno all’interno della stessacatena. Nel caso più frequente ciò avviene attraverso una rea-zione di backbiting nella quale la catena, ripiegandosi su se stes-sa, consente l’estrazione di un atomo di idrogeno da un’unitàmonomerica distante poche unità dall’estremità radicalica. Ilnumero di tali unità e il fatto stesso che tale reazione possa omeno avvenire dipendono dalla specifica struttura chimica dellacatena che ne determina la mobilità intramolecolare. In ognicaso questa reazione produce rami corti che si contraddistin-guono dai lunghi rami tipici della reazione di trasferimento inter-molecolare descritta precedentemente. Un tipico polimero incui i rami corti prevalgono su quelli lunghi è il polietilene abassa densità (Low Density PolyEthylene, LDPE). In questocaso i rami corti sono costituiti da non più di quattro o cinqueunità monomeriche e hanno una concentrazione superiore aquella dei rami lunghi di almeno un ordine di grandezza.
Reazione di b-scissione. La reazione di b-scissione si veri-fica a partire da un radicale localizzato in un qualche puntolungo la catena polimerica. Essa implica la scissione del lega-me C�C in posizione b rispetto al radicale, e porta alla for-mazione di una catena polimerica morta con doppio legameterminale e una catena radicalica attiva di dimensioni inferio-ri a quelle della catena originaria:
[18]
Considerando che la b-scissione avviene sempre dopo laformazione di un radicale interno alla catena, e dunque dopouna reazione di trasferimento a polimero o di backbiting, l’in-tero processo può essere visto come una reazione di trasferi-mento di catena, con la peculiarità di comportare la rotturadella macromolecola trasferita.
Questa reazione presenta ovviamente un’elevata energiadi attivazione e risulta determinante solo a temperature moltoelevate. Tipico è il caso della polimerizzazione in massa ad altatemperatura dello stirene, in cui la distribuzione dei pesi mole-colari degli oligomeri prodotti risulta controllata dalla reazio-ne di backbiting seguita da b-scissione.
Polimerizzazione radicalica controllata o pseudoviventeCome visto precedentemente, la caratteristica fondamentale
della cinetica della polimerizzazione a catena rispetto a quella a
kR
n
Rk
n
kfp
R RR R
R
kfp
R
kfm
RR
R
kfm
ASPETTI PROCESSISTICI
374 ENCICLOPEDIA DEGLI IDROCARBURI

stadi è che nella prima il tempo di vita delle catene radicalicheè assai minore della durata del processo di polimerizzazione.Ciò implica che alcune catene polimeriche vengono prodotteall’inizio e altre alla fine del processo, dove molte delle condi-zioni operative, quali la concentrazione delle diverse specie rea-genti, la viscosità e la temperatura, possono essere molto diver-se. Ciò può causare delle differenze tra le varie catene polime-riche in termini di lunghezza o di composizione, che comportanodisuniformità indesiderate nel prodotto finale. Questa situa-zione non si verifica nelle polimerizzazioni a stadi viste in pre-cedenza nelle quali le catene, cosiddette viventi, crescono tutteinsieme durante l’intero processo e risultano pertanto più unifor-mi. Il fatto che le catene siano viventi, e dunque rimangano reat-tive anche dopo il processo di sintesi, può comportare impor-tanti vantaggi nelle loro successive applicazioni, per esempioattraverso la realizzazione in situ di reticolazioni tra macromo-lecole e soprattutto la produzione di copolimeri a blocchi. Pertutti questi motivi risulta interessante poter condurre anche lereazioni radicaliche a catena in queste condizioni.
Poiché le reazioni di terminazione tra radicali sopra illu-strate sono irreversibili e certamente non eliminabili, l’obiet-tivo è in realtà raggiungere condizioni controllate o pseudovi-venti, in cui cioè le reazioni di terminazione siano fortementeridotte anche se non del tutto eliminate. Ciò viene ottenutointroducendo nel sistema reazioni di terminazione reversibilicon una specie capping, indicata con X, attraverso le quali lecatene radicaliche in crescita formano delle catene cosiddettedormienti, cioè non più in grado di propagare ma neppure diterminare. In questo caso ciascuna catena radicalica R puòseguire tre diverse reazioni: propagare con una molecola dimonomero, terminare in modo irreversibile con un altro radi-cale R o reagire in modo reversibile con la specie X per for-mare una catena dormiente. Poiché la terminazione bimoleco-lare è l’unica di ordine due rispetto alla concentrazione deiradicali, è possibile ridurne la velocità rispetto alle altre dueabbassando la concentrazione totale di radicali nel sistema. Ciòcomporta una concentrazione di catene dormienti, e dunqueviventi, superiore a quella delle catene morte e consente diapprossimare le condizioni di polimerizzazione vivente.
Tali condizioni vengono raggiunte scegliendo la concen-trazione e la reattività della specie X in modo da portare lamaggior parte dei radicali nel sistema sotto forma di speciedormienti. In questo caso il processo è molto lento, essendobassa la concentrazione di catene radicaliche che consumanomonomero, ma in compenso la formazione di catene morterisulta molto ridotta. Ciascuna catena cresce dunque durantenumerosissimi intervalli di tempo distribuiti all’interno del pro-cesso di polimerizzazione. Durante ciascuno di essi questaaddiziona poche unità monomeriche per poi tornare nello statodormiente dove la terminazione non può avvenire.
È opportuno osservare che questo meccanismo funzionasolo per ridurre l’effetto delle terminazioni bimolecolari, men-tre risulta inefficace nel ridurre le catene morte prodotte dallareazione di trasferimento a monomero.
La polimerizzazione vivente viene normalmente iniziataintroducendo nel sistema un iniziatore che contiene anche laspecie X, indicato con RX. Attraverso varie attivazioni e disat-tivazioni questa specie inserisce diverse (n) unità monomeri-che M dando luogo alle specie dormienti che hanno formaR�(Mn)�X.
Diversi sistemi chimici possono essere selezionati per rea-lizzare le condizioni sopra descritte, dando luogo a vari mec-canismi di polimerizzazione vivente.
Polimerizzazione mediata da nitrossido (Nitroxide Medi-ated Polymerization, NMP). Questo meccanismo di polime-rizzazione vivente consiste nella combinazione reversibile dellacatena vivente in crescita, R�n, e della cosiddetta specie radica-lica persistente, X� (per esempio il gruppo radicalico nitrossi-do), per formare la catena polimerica dormiente, RnX:
[19]
Varie possibilità per realizzare questo processo sono oggidisponibili, basate su diversi tipi di radicali persistenti. Tra que-sti il più usato è certamente il 2,2,6,6-tetrametilpiperidinil-1-ossi (TeMPO), che soffre peraltro della limitazione di esseredifficilmente applicabile a monomeri diversi dallo stirene e dirichiedere temperature di polimerizzazione relativamente ele-vate (120-140 °C).
Polimerizzazione radicalica per trasferimento atomico(Atom Transfer Radical Polymerization, ATRP). Questo mec-canismo è basato sulla reazione di addizione radicalica per tra-sferimento atomico ed è catalizzato da un metallo (tipicamen-te il rame): la rottura omolitica del legame in un alogenuroorganico avviene attraverso il trasferimento dell’alogeno alcomplesso metallico accompagnato dall’ossidazione del metal-lo. Il ciclo catalitico viene chiuso dalla restituzione dell’alo-geno al prodotto finale da parte del metallo di transizione. Èchiaro che, se il radicale prodotto può effettuare alcune rea-zioni di propagazione prima di dare luogo alla reazione inver-sa di trasferimento e se questo prodotto è ancora in grado didare luogo a un ciclo di trasferimento, questa reazione puòessere usata per produrre lo stesso scambio tra specie attive edormienti visto nel meccanismo precedente. La reazione rever-sibile risultante può essere rappresentata come segue:
[20]
dove X indica l’alogeno, Me(n) il metallo con stato di ossida-zione n e L il legante.
Il meccanismo ATRP deve il suo successo al fatto di esse-re applicabile a un’ampia gamma di monomeri, quali per esem-pio lo stirene, gli acrilati, i metacrilati, le acrilammidi e l’a-crilonitrile, che consentono a questa tecnica di produrre varitipi di polimeri a blocchi di notevole interesse applicativo. Lapresenza nel sistema di un metallo e di un legante relativamentecomplesso necessario a portare lo stesso in soluzione, nonchéla forte colorazione normalmente impartita da questo com-plesso al polimero, rappresentano i maggiori svantaggi di que-sto processo.
Trasferimento degenerativo (Degenerative Transfer, DT).In entrambi i meccanismi visti precedentemente lo scambio tralo stato attivo e quello dormiente è basato su una reazione(peraltro diversa) di terminazione reversibile. Conseguente-mente la reazione di scambio influisce sulla concentrazionetotale di radicali. Nelle polimerizzazioni radicaliche viventiper trasferimento degenerativo, la reazione di scambio avvie-ne invece per trasferimento diretto del gruppo terminale tra lacatena attiva e quella dormiente. Nel caso in cui venga utiliz-zato lo iodio come terminale di catena la reazione di scambiopuò essere schematizzata come segue:
[21]
Poiché questa reazione non cambia la concentrazione tota-le di radicali attivi, la concentrazione totale di catene termina-te per terminazione bimolecolare è uguale alla metà della con-centrazione iniziale di iniziatore. Pertanto la concentrazione ini-ziale delle specie che contengono iodio (nel seguito chiamate
R R I R R I� ���n m m n+ +➤||||||||
R X Me L R X Me L�nn
nn+ ++
6888( ) ( )/ /1
R X R X � �n n+ ������
PROCESSI DI POLIMERIZZAZIONE
375VOLUME V / STRUMENTI
����
����

semplicemente trasferitori di catena) determina il grado fina-le di polimerizzazione, nell’ipotesi in cui la concentrazione ini-ziale di iniziatore sia piccola rispetto alla concentrazione ini-ziale di trasferitore di catena.
Polimerizzazione per trasferimento reversibile di addizio-ne-frammentazione (Reversible Addition-Fragmentation Trans-fer, RAFT). Il processo RAFT può essere visto come un casoparticolare di trasferimento degenerativo. Il cosiddetto trasfe-ritore RAFT ha la struttura generale Q�Y�C(Z)�Y, doveY è lo zolfo, C il carbonio, Z è normalmente un gruppo feni-lico e Q è il gruppo vivente. La reazione procede attraversol’interazione tra una catena radicalica e una dormiente con laformazione di un intermedio di reazione dal quale la reazionepuò tornare indietro al radicale iniziale o procedere attraversoil trasferimento del gruppo Y�C(Z)�Y dalla specie dor-miente alla catena attiva:
[22]
La scelta corretta del gruppo Q è di fondamentale impor-tanza non solo perché questo risulta poi essere un gruppo ter-minale di catena (essendo l’altro terminale occupato dal grup-po RAFT), ma soprattutto perché esso determina la reattivitàiniziale dell’agente RAFT, influenzando in modo significati-vo la stabilità del radicale intermedio rispetto alla catena radi-calica attiva.
Sebbene i risultati più significativi siano stati ottenuti nelcaso della polimerizzazione RAFT dello stirene, il processorisulta assai efficiente per diverse altre specie monomeriche,come gli acrilati e i metacrilati. Inoltre questo processo risul-ta efficace a temperature moderate e può essere applicato consuccesso a sistemi di polimerizzazione in emulsione, nei qualiè possibile sfruttare la segregazione dei radicali in particellaper migliorare la produttività del processo. Un notevole svan-taggio di questo processo è la necessità di eliminare lo zolfodal prodotto di reazione, in quanto questo conferisce spesso alprodotto finale colorazioni non desiderate.
Catene non lineariIl meccanismo principale per la realizzazione di catene
polimeriche non lineari è basato sulla formazione di un radi-cale lungo una catena polimerica morta, dalla cui propagazio-ne si forma una cosiddetta ramificazione lunga. Ciò si verifi-ca attraverso la reazione di trasferimento a polimero (la reazio-ne di backbiting produce solo rami corti, non considerati in questocaso), la cui importanza aumenta al procedere della conversio-ne e dunque della concentrazione di catene morte. Un mecca-nismo alternativo è legato alla propagazione del doppio legameterminale che sfrutta la reattività residua lasciata all’interno della
catena morta da alcune reazioni di terminazione, e in partico-lare la terminazione per disproporzionamento, il trasferimen-to a monomero e la b-scissione. Anche in questo caso si rea-lizza lungo la catena un punto da cui si dipartono tre rami dilunghezza confrontabile, illustrati nella fig. 3 A.
Come già nel caso della polimerizzazione a stadi, la pro-cedura più efficace per produrre catene fortemente non linea-ri è basata sull’utilizzazione di monomeri che abbiano più diuna funzionalità reattiva, come nel caso dei dieni o dei mono-meri divilinici. In questo modo si ottengono catene polimeri-che morte che contengono però al loro interno svariati doppilegami, che possono propagare con altre catene radicaliche increscita attraverso reazioni di reticolazione. Come illustrato infig. 3 B, ciascuna di tali reazioni crea un punto di reticolazio-ne, cioè un centro da cui si dipartono quattro rami di dimen-sioni paragonabili.
La reazione di reticolazione è molto efficace nel far cre-scere il peso molecolare delle macromolecole e porta dunquefacilmente alla formazione di gel. Questo sistema si compor-ta in modo assai simile ai sistemi di polimerizzazione a stadi,in quanto le catene polimeriche rimangono reattive anche dopola reazione di terminazione a seguito della presenza dei doppilegami interni. Tuttavia la reticolazione in fig. 3B non è l’uni-ca reazione che possa portare alla formazione di gel nell’am-bito della polimerizzazione radicalica. Per ottenere un gel èinfatti necessario che le macromolecole crescano in modo espo-nenziale e dunque assai più rapido che non semplicementeaddizionando unità monomeriche, come nella propagazione.Questa condizione viene realizzata dalla reazione di reticola-zione, ma può essere raggiunta anche accoppiando due rea-zioni: una che riattivi le catene morte, come la reazione di tra-sferimento a polimero, e l’altra che le accoppi, come la termi-nazione bimolecolare per combinazione. Se la seconda vienesostituita dalla terminazione per disproporzionamento si pos-sono ottenere polimeri anche con elevatissimi gradi di ramifi-cazione ma non geli, in quanto viene a mancare il contributoalla crescita dato dalla reazione di terminazione per combina-zione.
6.4.4 Cinetica della polimerizzazioneradicalica
Nel seguito viene considerato un processo di polimerizzazio-ne radicalica con lo scopo di derivare opportune espressionicinetiche che ne descrivano l’evoluzione temporale in terminidi composizione e caratteristiche del polimero prodotto. Con-sideriamo un generico sistema in cui siano presenti tutte le rea-zioni elencate di seguito (insieme alle rispettive espressionidella velocità di reazione):
��➤|||||||| RR R YC(Z)Y�m n+
R YC Z YR R YC Z YR•� ��n m n m+ ( ) ( ) ➤||||||||
ASPETTI PROCESSISTICI
376 ENCICLOPEDIA DEGLI IDROCARBURI
A
B
� monomero
� monomero
fig. 3. Schematizzazione delle reazioni di propagazione del doppiolegame terminale (A) e di reticolazione (B).
����

[23] iniziazione: I R1 r�2fkdI�RI
propagazione: R�n�M Rn�1 r�kpMR�n
trasferimentiR�n�M R�1�Pn r�kfmMR�n
di catena: R�n�S R�1�Pn r�kfsSR�n
R�n�R�m Pn�m r�ktcR�nR�m
terminazioni: R�n�R�m Pn�Pm r�ktdR�nR�m
dove n, m�1,…,�
Si noti che con la lettera maiuscola si indicano sia il sim-bolo che (in corsivo) la concentrazione molare delle speciecoinvolte. In particolare, Rn e Pn rappresentano macromoleco-le contenenti n unità monomeriche radicaliche e morte, rispet-tivamente, M è il monomero, I l’iniziatore e S un trasferitoredi catena. Le reazioni riportate sono state discusse in prece-denza; si sono qui omesse le reazioni che portano alla forma-zione di ramificazioni, in modo da mantenere la trattazionelimitata al solo caso di catene lineari.
Le cinetiche di tutte le reazioni considerate sono del secon-do ordine, con l’unica eccezione della decomposizione del-l’iniziatore che è del prim’ordine. Per ciascuna di tali reazio-ni è possibile valutare un tempo caratteristico, significativodell’ordine di grandezza del tempo necessario alla reazioneper svilupparsi completamente nelle condizioni tipiche deiprocessi in esame. La valutazione di tali tempi consente diinquadrare in modo semplice e intuitivo le caratteristiche pecu-liari della cinetica dei processi di polimerizzazione radicali-ca. Risulta infatti che tali processi si svolgono attraverso rea-zioni caratterizzate da tre diversi livelli di tempi caratteristi-ci, i cui valori relativi determinano non solo la cineticadell’evoluzione del processo ma anche le caratteristiche delpolimero prodotto, come per esempio lunghezza e composi-zione delle catene.
Il tempo caratteristico del processo indica il tempo neces-sario al suo completamento, che nel caso in esame corrispon-de al consumo completo della specie monomerica. La velocitàdi consumo del monomero è data da:
[24]
dove R è la somma delle concentrazioni di tutti i radicali pre-senti nel sistema, R���
n�1R�n. È così possibile definire cometempo caratteristico di questo processo del secondo ordine lagrandezza:
[25]
dove kpR rappresenta la costante cinetica di pseudoprim’ordi-ne per la reazione di consumo del monomero. Nelle condizio-ni tipiche di questi processi il tempo caratteristico tM è del-l’ordine di 103-104 s.
Il tempo caratteristico tI della decomposizione dell’ini-ziatore, che è un processo del prim’ordine, è semplicementedato dall’inverso della costante cinetica del primo ordine, kd:
[26]
Poiché, come indicato precedentemente, il processo di poli-merizzazione per potersi sviluppare ha bisogno di una conti-nua produzione di radicali, questo tempo caratteristico deve
essere superiore, anche se non di molto, a quello del processo,cioè tI�tM.
Di un ordine di grandezza assai inferiore sono invece itempi caratteristici di tutte le reazioni che portano all’interru-zione della crescita della macromolecola per terminazione oper trasferimento. Questi sono i tempi caratteristici della rea-zione di terminazione per combinazione, ttc, e per dispropor-zionamento, ttd, e delle reazioni di trasferimento di catena amonomero, tfm, e al trasferitore di catena, tfs, che sono tuttidefiniti come i reciproci delle costanti cinetiche di pseudo-prim’ordine delle corrispondenti reazioni:
[27]
Per ogni specifico sistema di polimerizzazione solo alcu-ne di queste reazioni possono avvenire e con frequenze diver-se a seconda della chimica del sistema. In generale la reazio-ne che domina il processo di terminazione è quella che ha iltempo caratteristico più basso, che nei sistemi di più comuneinteresse è dell’ordine di 1 s. Questo determina il tempo chemediamente ciascun radicale ha a sua disposizione per propa-gare e addizionare monomeri prima che si verifichi la termi-nazione della catena. Ciò corrisponde dunque al tempo di vitadella catena che risulta, come precedentemente indicato, assaiinferiore al tempo caratteristico del processo, tM.
Il terzo livello di tempi caratteristici corrisponde ai pro-cessi più veloci, cioè quelli di addizione da parte del radicaledella singola unità monomerica. Come nel caso del tempo carat-teristico del processo, si fa riferimento alla reazione di propa-gazione, ma in questo caso si considera il processo di scom-parsa della specie radicalica. Il corrispondente tempo caratte-ristico è dunque definito da
[28]
che è dell’ordine di grandezza di 10�3-10�4 s.L’evoluzione cinetica di un processo di polimerizzazione
radicalica è basata sull’interazione di questi tre processi e inparticolare sui rapporti dei loro tempi caratteristici. Ciò deter-mina anche le caratteristiche delle catene polimeriche prodot-te. Per esempio, il numero di unità monomeriche mediamentecontenuto in una catena polimerica è dato dal rapporto tra iltempo caratteristico del processo di terminazione dominante,che come sopra menzionato è dell’ordine del secondo, e iltempo caratteristico del processo di propagazione. Affinché sipossa parlare di polimeri è necessario che tale rapporto siaalmeno dell’ordine di 100 o 1.000.
Tuttavia per una descrizione quantitativa più precisa dellacinetica del processo è necessario ricorrere ai bilanci materia-li delle specie reagenti, considerando per ciascuna specie tuttele reazioni in cui essa è coinvolta. La forma che tali bilanci assu-mono dipende però dal tipo di reattore che viene considerato eciò dilaterebbe notevolmente la trattazione. Nel seguito si faràdunque riferimento ai più comuni reattori discontinui (batch)in cui i reagenti vengono introdotti tutti all’inizio del processo,senza ulteriori aggiunte o prelievi durante la reazione.
Consumo del monomeroIl monomero viene principalmente consumato dalle catene
radicaliche in crescita presenti nel reattore. Il primo passo è dun-que la valutazione della concentrazione globale R di tali catene,indipendentemente dalla loro lunghezza. Tale concentrazione puòessere valutata dal bilancio materiale globale delle catene attive:
τ ppk M
= 1
τ τ τ τtctc
tdtd
fmfm
fsfk R k R k M k
= = = =1 1 1 1
ssS
τ Idk
= 1
τMpk R
= 1
ddMt
k RMp= −
����➤||||||||ktd
����➤||||||||ktc
����➤||||||||k fs
����➤||||||||k fm
����➤||||||||kp
����➤||||||||
PROCESSI DI POLIMERIZZAZIONE
377VOLUME V / STRUMENTI

[29]
dove kt�ktc�ktd è la costante cinetica complessiva delle rea-zioni di terminazione bimolecolare e RI rappresenta la velocitàdi produzione di radicali. Nel caso in cui questi siano prodot-ti dalla decomposizione di un iniziatore I come indicato dallaprima delle reazioni [23], si ha:
[30]
La concentrazione dell’iniziatore, I, viene a sua volta otte-nuta da un bilancio materiale che nel caso più semplice di reat-tore isotermo porta all’espressione analitica
[31]
dove I0 rappresenta la concentrazione iniziale di iniziatore.Come già evidenziato, l’aspetto fondamentale dei processi
di polimerizzazione radicalica a catena è che il tempo caratte-ristico di terminazione delle catene è molto breve rispetto a quel-lo della loro produzione, che è confrontabile con quello di decom-posizione dell’iniziatore, tI. Ciò consente di applicare al lorobilancio [29] l’approssimazione di pseudostazionarietà secon-do la quale il termine di accumulo, corrispondente alla deriva-ta temporale, può essere trascurato rispetto ai termini di gene-razione e consumo. Ciò riduce la [29] a una relazione algebri-ca che consente di valutare la concentrazione di radicali come
[32]
dove kt�ktc�ktd. A questo punto è possibile considerare il bilan-cio materiale per la specie monomerica che, facendo sempreriferimento a un reattore batch isotermo, si riduce a
[33]
Considerando che la velocità di produzione di radicali RIsi mantenga costante durante il processo e che pure le costan-ti cinetiche ktc e kp non varino, la relazione precedente può esse-re integrata analiticamente portando alla tipica espressioneesponenziale per la concentrazione del monomero:
[34]
dove M0 rappresenta la concentrazione iniziale di monomero. Da questa relazione risulta che, per un assegnato siste-
ma di iniziazione (e dunque del valore di RI), la cinetica diconsumo del monomero è determinata unicamente dal rap-porto kp ��
1
kt . Ciò implica altresì che dalla misura della velo-cità di consumo di monomero è possibile stimare al più ilvalore di tale rapporto, ma non il valore assoluto delle sin-gole costanti cinetiche. Conseguentemente è frequente tro-vare in letteratura i valori del rapporto kp ��
1
kt per varie spe-cie monomeriche. I valori assoluti delle costanti cinetichepossono essere stimati con tecniche più complesse, come peresempio la polimerizzazione a laser pulsato, che richiede lacaratterizzazione della lunghezza delle catene polimericheprodotte.
L’espressione [34] rappresenta nella pratica solo un’ap-prossimazione, in quanto i termini del bilancio [33] variano neltempo. Particolarmente sensibile è l’effetto della variazionedella viscosità del sistema che, attraverso l’effetto Trommsdorff,produce una diminuzione della costante di terminazione per
combinazione e dunque un rapido aumento della concentra-zione di radicali e quindi della velocità di consumo del mono-mero. A questo effetto può sommarsi quello dovuto alla dimi-nuzione del coefficiente di scambio termico del reattore, cau-sata dall’aumentare della viscosità del sistema reagente, cheporta a una diminuzione della velocità di smaltimento del calo-re prodotto e dunque a un aumento della temperatura, che acce-lera ulteriormente il processo di consumo del monomero. Inconclusione i profili di concentrazione di monomero all’in-terno del reattore deviano dalla forma esponenziale ideale,come illustrato nella fig. 4.
Distribuzione della lunghezza delle cateneLe caratteristiche applicative di un polimero sono deter-
minate dalla composizione e dalla struttura delle macromo-lecole che lo compongono. Come si vedrà di seguito, le macro-molecole non sono tutte identiche ma costituiscono in gene-rale una popolazione di individui con caratteristiche diverse;è la distribuzione di tali caratteristiche, e non solo i relativivalori medi, che definisce le caratteristiche applicative di unmateriale polimerico. Risulta pertanto particolarmente inte-ressante descrivere la cinetica attraverso cui tali distribuzio-ni evolvono in funzione delle particolari condizioni opera-tive del processo di polimerizzazione. A questo scopo siutilizzano particolari bilanci materiali, detti bilanci di popo-lazione, che sono riferiti a specifici individui di queste popo-lazioni e in particolare a quelli caratterizzati da un partico-lare valore dell’elemento caratteristico considerato. Nel casoin cui quest’ultimo sia la lunghezza della catena polimerica(detto anche grado di polimerizzazione, che rappresenta ilnumero di unità monomeriche contenute nella catena), i cor-rispondenti bilanci di popolazione consentono di valutare ladistribuzione della lunghezza della catena (Chain LengthDistribution, CLD).
La CLD delle catene polimeriche morte presenti in un reat-tore discontinuo a fine processo può essere valutata attraver-so i seguenti passi: valutazione della CLD istantanea, cioè laCLD delle catene prodotte a un certo istante durante il corsodel processo di polimerizzazione, e cumulazione di tutte leCLD istantanee, pesate sulle corrispondenti quantità di poli-mero prodotto, per valutare la CLD cumulata, cioè quella rela-tiva al prodotto finale (v. anche cap. 8.1).
È opportuno ricordare che tale procedura è possibile gra-zie alla caratteristica peculiare di questi processi per cui le cate-ne vengono prodotte in intervalli di tempo brevissimi rispettoalla durata del processo.
M M kRk
tpI
t
= −
0
exp
ddMt
k MRp= −
RRk
I
t
=
I I k td= −( )0exp
R fk II d= 2
ddRt
R k RI t= − 2
ASPETTI PROCESSISTICI
378 ENCICLOPEDIA DEGLI IDROCARBURI
1
eq. [34]
effetto Trommsdorff
tempo
conv
ersi
one
fig. 4. Ruolo dell’effetto Trommsdorff sull’andamento della conversione in funzione del tempo in un reattore batch.
La distribuzione istantanea numerale delle lunghezze dicatena, fN(n), tale per cui fN(n)dn rappresenta la frazione nume-rica di catene polimeriche di lunghezza n prodotte a un certoistante, è data dalla relazione
[35]
dove sono stati introdotti due nuovi parametri cinetici adi-mensionali, definiti come rapporti tra tempi caratteristici:
[36]
[37]
con a�b�g. Dalle relazioni sopra riportate è possibile osser-vare che i parametri cinetici b e g influenzano in modo diversola CLD istantanea, pur essendo verificato che all’aumentare dientrambi si ottengono catene mediamente più corte. Ciò è ragio-nevole in quanto valori più elevati di tali parametri corrispon-dono a valori più elevati delle terminazioni rispetto alla propa-gazione. È interessante osservare che i parametri b e g raggrup-pano i tempi caratteristici di processi di terminazione che sonoqualitativamente diversi tra loro. Nel primo si trova la termina-zione per combinazione in cui le catene che terminano perdonola propria identità attraverso il processo di terminazione, for-mando un’unica catena che ha per lunghezza la somma di quel-le delle due originarie. In g invece sono compresi tre processi diterminazione che sono simili tra loro in quanto mantengono inal-terata l’identità della catena attraverso la terminazione. Tale dif-ferenza strutturale delle terminazioni fa sì che queste agiscanoin modo differente sulla CLD e che dunque i rispettivi parame-tri cinetici compaiano in diversi parametri adimensionali.
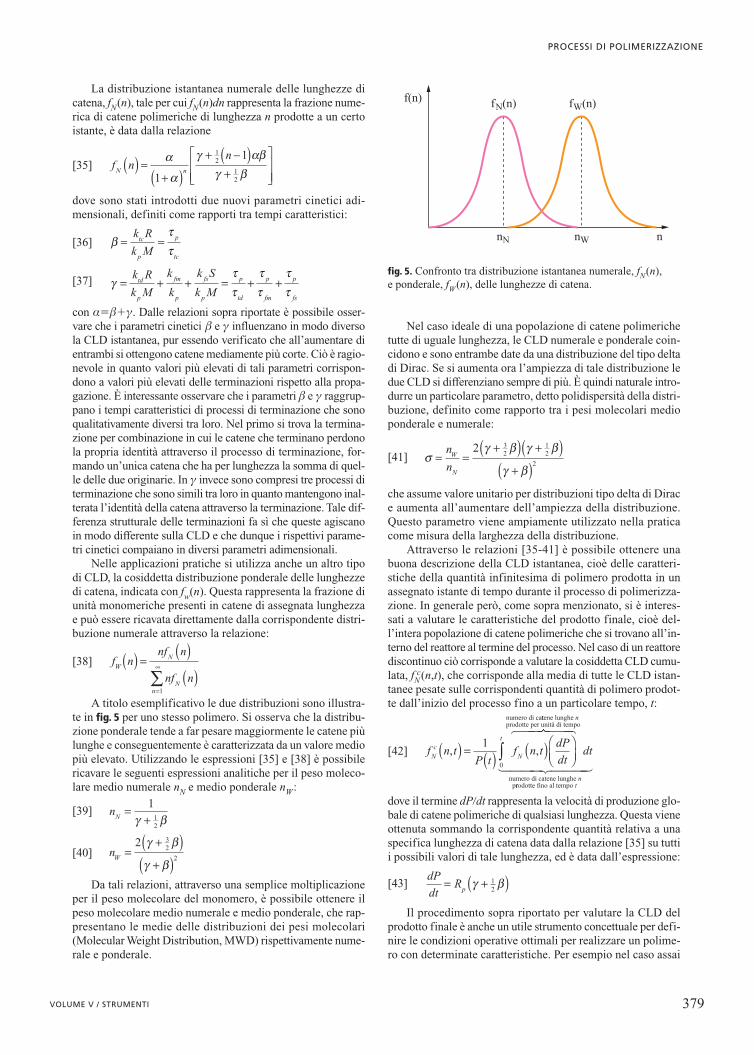
Nelle applicazioni pratiche si utilizza anche un altro tipodi CLD, la cosiddetta distribuzione ponderale delle lunghezzedi catena, indicata con fw(n). Questa rappresenta la frazione diunità monomeriche presenti in catene di assegnata lunghezzae può essere ricavata direttamente dalla corrispondente distri-buzione numerale attraverso la relazione:
[38]
A titolo esemplificativo le due distribuzioni sono illustra-te in fig. 5 per uno stesso polimero. Si osserva che la distribu-zione ponderale tende a far pesare maggiormente le catene piùlunghe e conseguentemente è caratterizzata da un valore mediopiù elevato. Utilizzando le espressioni [35] e [38] è possibilericavare le seguenti espressioni analitiche per il peso moleco-lare medio numerale nN e medio ponderale nW:
[39]
[40]
Da tali relazioni, attraverso una semplice moltiplicazioneper il peso molecolare del monomero, è possibile ottenere ilpeso molecolare medio numerale e medio ponderale, che rap-presentano le medie delle distribuzioni dei pesi molecolari(Molecular Weight Distribution, MWD) rispettivamente nume-rale e ponderale.
Nel caso ideale di una popolazione di catene polimerichetutte di uguale lunghezza, le CLD numerale e ponderale coin-cidono e sono entrambe date da una distribuzione del tipo deltadi Dirac. Se si aumenta ora l’ampiezza di tale distribuzione ledue CLD si differenziano sempre di più. È quindi naturale intro-durre un particolare parametro, detto polidispersità della distri-buzione, definito come rapporto tra i pesi molecolari medioponderale e numerale:
[41]
che assume valore unitario per distribuzioni tipo delta di Dirace aumenta all’aumentare dell’ampiezza della distribuzione.Questo parametro viene ampiamente utilizzato nella praticacome misura della larghezza della distribuzione.
Attraverso le relazioni [35-41] è possibile ottenere unabuona descrizione della CLD istantanea, cioè delle caratteri-stiche della quantità infinitesima di polimero prodotta in unassegnato istante di tempo durante il processo di polimerizza-zione. In generale però, come sopra menzionato, si è interes-sati a valutare le caratteristiche del prodotto finale, cioè del-l’intera popolazione di catene polimeriche che si trovano all’in-terno del reattore al termine del processo. Nel caso di un reattorediscontinuo ciò corrisponde a valutare la cosiddetta CLD cumu-lata, fN
c(n,t), che corrisponde alla media di tutte le CLD istan-tanee pesate sulle corrispondenti quantità di polimero prodot-te dall’inizio del processo fino a un particolare tempo, t:
[42]
dove il termine dP�dt rappresenta la velocità di produzione glo-bale di catene polimeriche di qualsiasi lunghezza. Questa vieneottenuta sommando la corrispondente quantità relativa a unaspecifica lunghezza di catena data dalla relazione [35] su tuttii possibili valori di tale lunghezza, ed è data dall’espressione:
[43]
Il procedimento sopra riportato per valutare la CLD delprodotto finale è anche un utile strumento concettuale per defi-nire le condizioni operative ottimali per realizzare un polime-ro con determinate caratteristiche. Per esempio nel caso assai
ddPt
Rp= +( )γ β1
2
f n tP t
f n t PtN
cN, ,( ) = ( ) ( )
1 dd
numero di cattene lunghe prodotte per unità di tempo
n
� ���� ���
dtt
n
0
∫ numero di catene lunghe prrodotte fino al tempo t
� ��� ���
σγ β γ β
γ β= =
+( ) +( )+( )
nn
W
N
2 3
2
1
2
2
nW =+( )+( )
2 3
2
2
γ β
γ β
nN =+1
1
2γ β
f nnf n
nf nW
N
Nn
( ) =( )
( )=
∞
∑1
γττ
ττ
ττ
= + + = + +k Rk M
kk
k Sk M
td
p
fm
p
fs
p
p
td
p
fm
p
fs
βττ
= =k Rk M
tc
p
p
tc
f nn
N n( ) =+( )
+ −( )+
α
α
γ αβγ β1
11
2
1
2
PROCESSI DI POLIMERIZZAZIONE
379VOLUME V / STRUMENTI
nN nW n
fN(n) fW(n)f(n)
fig. 5. Confronto tra distribuzione istantanea numerale, fN(n), e ponderale, fW(n), delle lunghezze di catena.
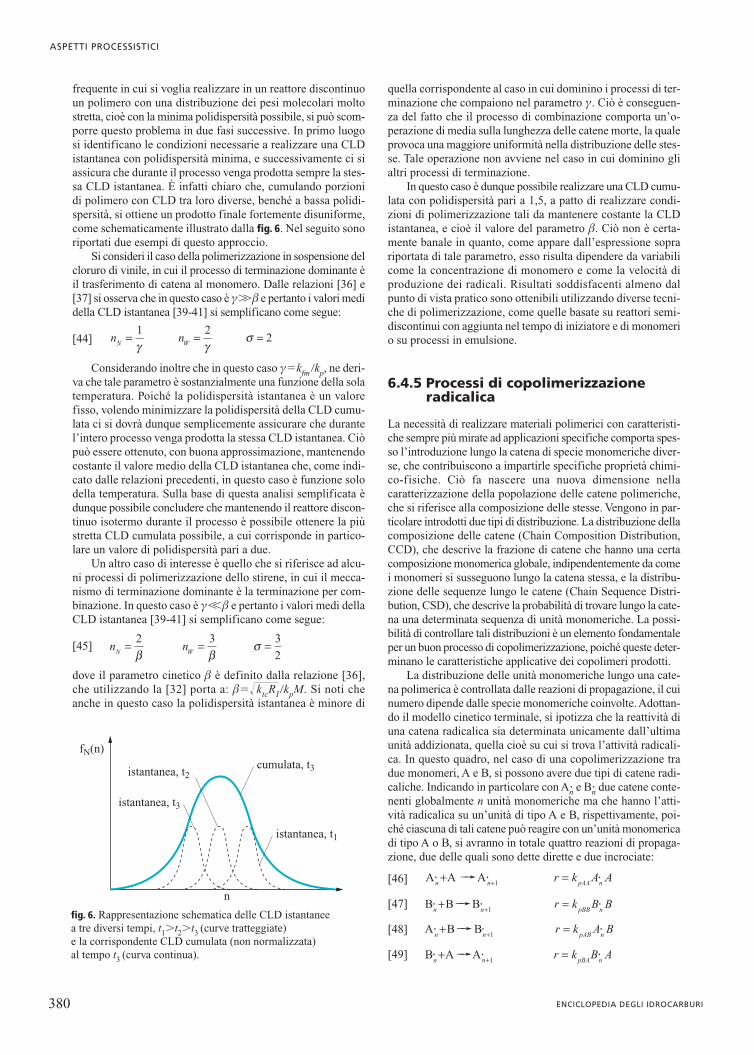
frequente in cui si voglia realizzare in un reattore discontinuoun polimero con una distribuzione dei pesi molecolari moltostretta, cioè con la minima polidispersità possibile, si può scom-porre questo problema in due fasi successive. In primo luogosi identificano le condizioni necessarie a realizzare una CLDistantanea con polidispersità minima, e successivamente ci siassicura che durante il processo venga prodotta sempre la stes-sa CLD istantanea. È infatti chiaro che, cumulando porzionidi polimero con CLD tra loro diverse, benché a bassa polidi-spersità, si ottiene un prodotto finale fortemente disuniforme,come schematicamente illustrato dalla fig. 6. Nel seguito sonoriportati due esempi di questo approccio.
Si consideri il caso della polimerizzazione in sospensione delcloruro di vinile, in cui il processo di terminazione dominante èil trasferimento di catena al monomero. Dalle relazioni [36] e[37] si osserva che in questo caso è g��b e pertanto i valori medidella CLD istantanea [39-41] si semplificano come segue:
[44]
Considerando inoltre che in questo caso g�kfm �kp, ne deri-va che tale parametro è sostanzialmente una funzione della solatemperatura. Poiché la polidispersità istantanea è un valorefisso, volendo minimizzare la polidispersità della CLD cumu-lata ci si dovrà dunque semplicemente assicurare che durantel’intero processo venga prodotta la stessa CLD istantanea. Ciòpuò essere ottenuto, con buona approssimazione, mantenendocostante il valore medio della CLD istantanea che, come indi-cato dalle relazioni precedenti, in questo caso è funzione solodella temperatura. Sulla base di questa analisi semplificata èdunque possibile concludere che mantenendo il reattore discon-tinuo isotermo durante il processo è possibile ottenere la piùstretta CLD cumulata possibile, a cui corrisponde in partico-lare un valore di polidispersità pari a due.
Un altro caso di interesse è quello che si riferisce ad alcu-ni processi di polimerizzazione dello stirene, in cui il mecca-nismo di terminazione dominante è la terminazione per com-binazione. In questo caso è gb e pertanto i valori medi dellaCLD istantanea [39-41] si semplificano come segue:
[45]
dove il parametro cinetico b è definito dalla relazione [36],che utilizzando la [32] porta a: b��
14422
ktcRI �kpM. Si noti cheanche in questo caso la polidispersità istantanea è minore di
quella corrispondente al caso in cui dominino i processi di ter-minazione che compaiono nel parametro g. Ciò è conseguen-za del fatto che il processo di combinazione comporta un’o-perazione di media sulla lunghezza delle catene morte, la qualeprovoca una maggiore uniformità nella distribuzione delle stes-se. Tale operazione non avviene nel caso in cui dominino glialtri processi di terminazione.
In questo caso è dunque possibile realizzare una CLD cumu-lata con polidispersità pari a 1,5, a patto di realizzare condi-zioni di polimerizzazione tali da mantenere costante la CLDistantanea, e cioè il valore del parametro b. Ciò non è certa-mente banale in quanto, come appare dall’espressione soprariportata di tale parametro, esso risulta dipendere da variabilicome la concentrazione di monomero e come la velocità diproduzione dei radicali. Risultati soddisfacenti almeno dalpunto di vista pratico sono ottenibili utilizzando diverse tecni-che di polimerizzazione, come quelle basate su reattori semi-discontinui con aggiunta nel tempo di iniziatore e di monomerio su processi in emulsione.
6.4.5 Processi di copolimerizzazioneradicalica
La necessità di realizzare materiali polimerici con caratteristi-che sempre più mirate ad applicazioni specifiche comporta spes-so l’introduzione lungo la catena di specie monomeriche diver-se, che contribuiscono a impartirle specifiche proprietà chimi-co-fisiche. Ciò fa nascere una nuova dimensione nellacaratterizzazione della popolazione delle catene polimeriche,che si riferisce alla composizione delle stesse. Vengono in par-ticolare introdotti due tipi di distribuzione. La distribuzione dellacomposizione delle catene (Chain Composition Distribution,CCD), che descrive la frazione di catene che hanno una certacomposizione monomerica globale, indipendentemente da comei monomeri si susseguono lungo la catena stessa, e la distribu-zione delle sequenze lungo le catene (Chain Sequence Distri-bution, CSD), che descrive la probabilità di trovare lungo la cate-na una determinata sequenza di unità monomeriche. La possi-bilità di controllare tali distribuzioni è un elemento fondamentaleper un buon processo di copolimerizzazione, poiché queste deter-minano le caratteristiche applicative dei copolimeri prodotti.
La distribuzione delle unità monomeriche lungo una cate-na polimerica è controllata dalle reazioni di propagazione, il cuinumero dipende dalle specie monomeriche coinvolte. Adottan-do il modello cinetico terminale, si ipotizza che la reattività diuna catena radicalica sia determinata unicamente dall’ultimaunità addizionata, quella cioè su cui si trova l’attività radicali-ca. In questo quadro, nel caso di una copolimerizzazione tradue monomeri, A e B, si possono avere due tipi di catene radi-caliche. Indicando in particolare con A�n e B�n due catene conte-nenti globalmente n unità monomeriche ma che hanno l’atti-vità radicalica su un’unità di tipo A e B, rispettivamente, poi-ché ciascuna di tali catene può reagire con un’unità monomericadi tipo A o B, si avranno in totale quattro reazioni di propaga-zione, due delle quali sono dette dirette e due incrociate:
[46]
[47]
[48]
[49] B A A� � ���n n pBA nr k B A+ =+➤||||||||
1
A B B� � ���n n pAB nr k A B+ =+➤||||||||
1
B B B� � ���n n pBB nr k B B+ =+➤||||||||
1
A A A� � ���n n pAA nr k A A+ =+➤||||||||
1
n nN W= = =2 3 3
2β βσ
n nN W= = =1 22
γ γσ
ASPETTI PROCESSISTICI
380 ENCICLOPEDIA DEGLI IDROCARBURI
fN(n)
istantanea, t1
cumulata, t3
istantanea, t3
istantanea, t2
n
fig. 6. Rappresentazione schematica delle CLD istantaneea tre diversi tempi, t1�t2�t3 (curve tratteggiate) e la corrispondente CLD cumulata (non normalizzata)al tempo t3 (curva continua).

A fianco di ciascuna delle reazioni di propagazione è indi-cata la corrispondente espressione della velocità di reazione.In accordo al modello cinetico terminale le velocità sono descrit-te da cinetiche del secondo ordine, le cui costanti cinetichesono identificate esclusivamente dal tipo di unità in cui si troval’attività radicalica sulla catena reagente e dal tipo di speciemonomerica coinvolta.
Oltre alle reazioni di propagazione in questi processi sonopresenti anche le reazioni di iniziazione, terminazione e tra-sferimento di catena, che sono analoghe a quelle presenti nellaomopolimerizzazione ma in numero superiore in ragione delpiù elevato numero di tipi di radicali presenti.
Un modello cinetico più accurato, il cosiddetto modellocinetico penultimo, consente di tenere conto dell’effetto dellapenultima unità monomerica sulla reattività del radicale. Ciòcomporta naturalmente un aumento del numero di reazioni dipropagazione possibili, in quanto ciascuna delle reazioni [46-49] avviene con velocità diversa a seconda che l’unità mono-merica che precede quella su cui si trova l’attività radicalicasia di tipo A o B. Questo modello viene scarsamente utilizza-to in pratica, anche in considerazione dei buoni risultati gene-ralmente forniti dal modello cinetico terminale.
Distribuzione della lunghezza delle cateneLa prima informazione necessaria per descrivere la cine-
tica di crescita delle catene di copolimero riguarda la compo-sizione della miscela reagente in termini di catene radicalicheattive. In particolare è necessario conoscere la frazione di cate-ne che terminano con ciascuna delle unità monomeriche pre-senti in quanto, secondo il modello cinetico terminale adotta-to, questa ne definisce la reattività.
A questo scopo è necessario considerare i bilanci materia-li delle specie radicaliche, differenziandole solo sulla base deltipo di unità radicalica. In questi bilanci dovranno essere presein considerazione tutte le reazioni che comportano la scom-parsa o la produzione di un radicale di un certo tipo. Oltre allereazioni di iniziazione e terminazione, che causano la forma-zione o la eliminazione di una specie radicalica, dovranno dun-que essere considerate anche le reazioni di propagazione incro-ciata, in quanto queste modificano il tipo di unità radicalica.Considerando anzi che tali reazioni sono generalmente assaipiù veloci di quelle di terminazione e iniziazione, queste ulti-me vengono generalmente trascurate nell’ambito della cosid-detta approssimazione di catena lunga. Si consideri a titoloesemplificativo il caso di una polimerizzazione con tre mono-meri, A, B e C. Indicando con A�, B� e C� la concentrazioneglobale di catene radicaliche che terminano con ciascuna delletre unità monomeriche, i bilanci di cui sopra assumono la forma
[50]
[51]
[52]
Sommando le equazioni precedenti membro a membro siottiene che la variazione temporale della concentrazione totaledi radicali, R�A��B��C�, è uguale a zero. Ciò non è ovvia-mente corretto e riflette il fatto che questi bilanci sono stati scrit-ti nell’ambito della approssimazione di catena lunga. Essi con-sentono infatti di valutare solo la composizione della miscela di
radicali e non i valori assoluti delle loro concentrazioni. Intro-ducendo le frazioni molari pi di catene radicaliche di ogni tipo:
[53]
e sostituendo nelle equazioni [50] e [51], in cui i termini diaccumulo vengono trascurati secondo la approssimazione dipseudostazionarietà, si ottiene
[54]
[55]
[56]
Si noti che l’ultima relazione di consistenza viene aggiuntaper chiudere il sistema, la cui soluzione fornisce la desideratacomposizione del sistema reagente in termini di catene con diver-sa attività radicalica. Tale sistema può essere facilmente gene-ralizzato a un numero qualsiasi di specie monomeriche e in par-ticolare nel caso binario produce la seguente soluzione:
[57]
Queste relazioni sono valide nell’ambito dell’approssima-zione di catena lunga che può essere applicata con ottima accu-ratezza a tutti i copolimeri con esclusione dei copolimeri a bloc-chi. Questi sono costituiti da poche lunghissime sequenze omo-polimeriche, che corrispondono al fatto che le propagazioniincrociate sono molto più lente di quelle dirette, tanto da esse-re confrontabili a terminazioni e iniziazioni. In questo caso nonè evidentemente possibile trascurare queste ultime nei bilanci[50-52].
La descrizione del comportamento cinetico di un sistemache coinvolga molte specie monomeriche appare certamenteassai complicata per l’elevato numero di specie reagenti coin-volte. Tuttavia, nell’ambito delle approssimazioni sopra descrit-te, è possibile derivare una soluzione analitica molto sempli-ce che viene indicata come approccio pseudocinetico. Essoconsiste nell’utilizzare le stesse relazioni derivate preceden-temente per descrivere la cinetica del processo di omopoli-merizzazione, sostituendo le costanti cinetiche delle reazionicoinvolte con opportune costanti pseudocinetiche. Quest’ul-time sono ottenute a partire dalle costanti cinetiche reali delprocesso di copolimerizzazione attraverso opportune mediepesate sulle frazioni molari delle diverse specie radicalichenel sistema.
Tali medie sono diverse a seconda dell’ordine della rea-zione rispetto alle specie radicaliche. In particolare, con rife-rimento a un sistema ternario, si ha per la reazione di propa-gazione dei monomeri A, B e C:
[58]
[59]
[60]
da cui si ricava la costante pseudocinetica globale di propaga-zione, k*
p, attraverso una media pesata sulle frazioni molari deimonomeri, xA, xB e xC:
[61]
Per le reazioni di trasferimento di catena si hanno le seguen-ti espressioni delle costanti pseudocinetiche:
k k x k x k xp pA A pB B pC C∗ ∗ ∗ ∗= + +
k k p k p k ppC pAC A pBC B pCC C∗ = + +
k k p k p k ppB pAB A pBB B pCB C∗ = + +
k k p k p k ppA pAA A pBA B pCA C∗ = + +
pk A
k A k Bp
k Bk A k BA
pBA
pBA pABB
pAB
pBA pAB
=+
=+
p p pA B C+ + = 1
k Bp k Bp k A k C ppAB A pCB C pBA pBC B+ − +( ) = 0
k Ap k Ap k B k C ppBA B pCA C pAB pAC A+ − +( ) = 0
p AR
p BR
p CRA B C= = =� � �
ddCt
k A C k B C k A k B CpAC pBC pCA pCB
�� � �= + − +( )
ddBt
k A B k C B k A k C BpAB pCB pBA pBC
�� � �= + − +( )
ddAt
k B A k C A k B k C ApBA pCA pAB pAC
�� � �= + − +( )
PROCESSI DI POLIMERIZZAZIONE
381VOLUME V / STRUMENTI

[62]
[63]
che sono diverse per le reazioni di terminazione in quanto que-ste sono di secondo ordine rispetto alle specie radicaliche:
[64]
[65]
Nell’ambito del modello pseudocinetico è possibile dun-que valutare la concentrazione totale R di radicali utilizzandola stessa equazione [32] ma sostituendo le costanti cinetichedi omopolimerizzazione con quelle pseudocinetiche corri-spondenti:
[66]
Da questa è poi possibile valutare la velocità di consumodella singola specie monomerica, come per esempio quellarelativa al monomero A:
[67]
Anche la CLD di un copolimero può essere valutata median-te le stesse relazioni [35-41] valide per l’omopolimero, utiliz-zando per la valutazione dei parametri b e g le corrisponden-ti costanti pseudocinetiche. In questo caso la variabile M rap-presenta la concentrazione totale di tutte le specie monomerichenel sistema.
Distribuzione della composizione delle cateneLa distribuzione delle diverse specie monomeriche lungo
la catena polimerica è determinata dalla probabilità che il sin-golo radicale ha di addizionare l’una o l’altra delle specie mono-meriche presenti. A sua volta questa probabilità è definita dallareattività intrinseca del singolo monomero e dalla sua con-centrazione. In generale i diversi monomeri hanno reattivitàdiversa e dunque si consumano in modo diverso durante il pro-cesso di polimerizzazione, con la conseguenza che la compo-sizione monomerica varia nel tempo. Poiché il tempo di vitadelle catene è molto più breve di quello del processo, si avran-no catene prodotte a istanti diversi lungo il processo, e quindipresumibilmente con composizione diversa in quanto prodot-te a partire da differenti composizioni della miscela monome-rica. Questo processo, che viene chiamato usualmente deriva(o drift) compositiva, rappresenta spesso un problema impor-tante dal punto di vista applicativo, in quanto dà luogo a pro-dotti finali molto disuniformi, nel senso di catene aventi com-posizione molto diversa tra loro.
Per studiare quantitativamente questo processo è necessa-rio valutare la composizione media di una catena polimerica.Poiché durante la vita di una catena polimerica il consumo dimonomero e quindi gli eventuali cambiamenti di composizio-ne sono trascurabili, possiamo valutare la composizione dellacatena a partire dalla velocità istantanea di consumo delle sin-gole specie monomeriche. Così, per esempio nel caso di duespecie monomeriche A e B, la frazione molare FA di monome-ro A, presente all’interno di una catena copolimerica prodottaa un certo istante, è data dal rapporto:
[68]
dove RpA e RpB rappresentano le velocità di consumo delle diver-se specie monomeriche valutate mediante la [67] a quello stes-so istante. Attraverso opportune rielaborazioni è possibile riscri-vere la [68] nella forma
[69]
Tale relazione indica che la composizione istantanea dellacatena polimerica è determinata dalla frazione molare dellespecie monomeriche, xA e xB, e da due parametri cinetici adi-mensionali:
[70]
che vengono detti rapporti di reattività e rappresentano per cia-scun tipo di radicale il rapporto tra la costante cinetica di pro-pagazione diretta e incrociata.
Della relazione [69] viene normalmente data una rappre-sentazione grafica in termini del diagramma di Mayo e Lewis.Come illustrato in fig. 7 questo diagramma rappresenta la com-posizione istantanea delle catene polimeriche FA in funzionedella composizione monomerica xA della miscela da cui que-ste vengono generate. La diagonale corrisponde al caso in cuii monomeri hanno uguale reattività, cioè rA�1 e rB�1 nella[69], e dunque le catene polimeriche hanno la stessa compo-sizione della miscela monomerica da cui provengono, cioèFA�xA. In generale però i monomeri hanno reattività diversa eil corrispondente diagramma può assumere vari andamenti,come quelli illustrati dalle diverse curve in fig. 7. Per esempionel caso in cui il monomero A sia più reattivo nei confronti dientrambe le specie radicaliche, si ha rA�1 e rB1 e dunque lecatene polimeriche sono più ricche nel monomero A dellamiscela monomerica da cui derivano. Al contrario, nel caso sia
rkk
rkkA
pAA
pABB
pBB
pBA
= =
Fr x x x
r x x x r x x xAA A B A
A A B A B B A B
=+( )
+( ) + +( )
FR
R RApA
pA pB
=+
R k ARpA pA= ∗
RR
k kI
tc td
=+∗ ∗
k k p k p k pk
td tdAA A tdBB B tdCC C
t
∗ = + ++
2 2 2
2 ddAB A B tdAC A C tdBC B Cp p k p p k p p+ +2 2
k k p k p k pk
tc tcAA A tcBB B tcCC C
t
∗ = + ++
2 2 2
2 ccAB A B tcAC A C tcBC B Cp p k p p k p p+ +2 2
k k p k p k pfs fsA A fsB B fsC C∗ = + +
k k p k p k pfm fmA A fmB B fmC C∗ = + +
ASPETTI PROCESSISTICI
382 ENCICLOPEDIA DEGLI IDROCARBURI
FA
xA 10
III
II
I
0
1
fig. 7. Diagramma di Mayo e Lewis che rappresenta la frazione molare istantanea FA del monomero A nel polimero in funzione della frazione molare xA dello stessonella miscela reagente di monomeri per tre diverse coppie di valori dei rapporti di reattività: rA�1 e rB1 (curva I),rA1 e rB�1 (curva II) e rA1 e rB1 (curva III).
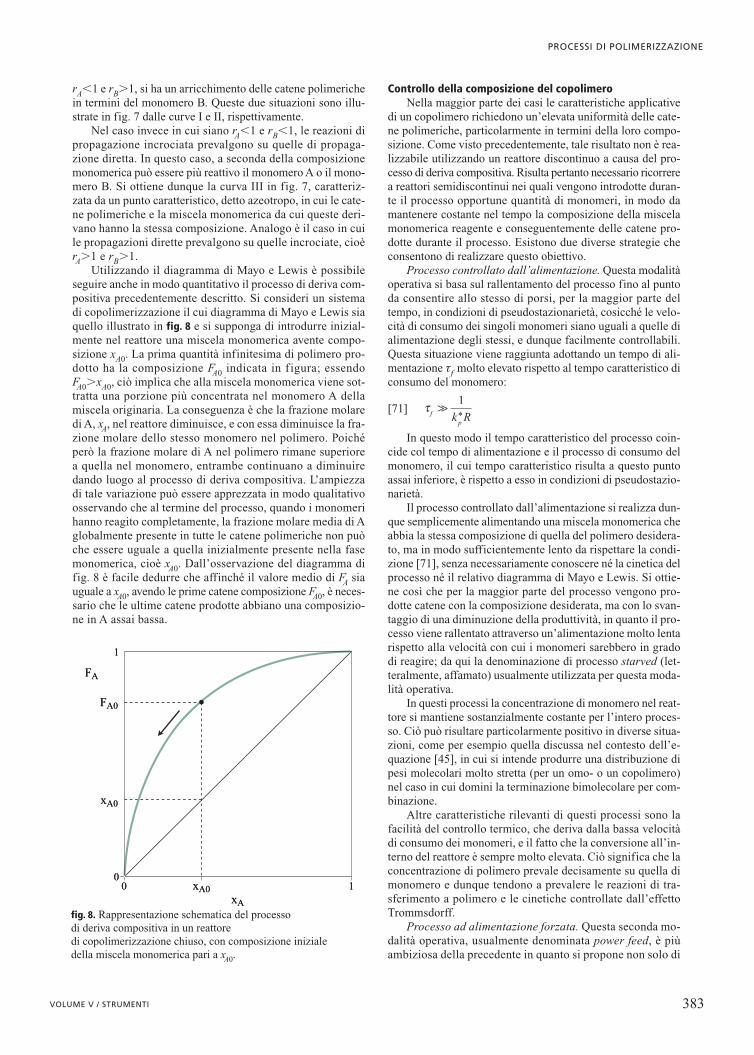
rA1 e rB�1, si ha un arricchimento delle catene polimerichein termini del monomero B. Queste due situazioni sono illu-strate in fig. 7 dalle curve I e II, rispettivamente.
Nel caso invece in cui siano rA1 e rB1, le reazioni dipropagazione incrociata prevalgono su quelle di propaga-zione diretta. In questo caso, a seconda della composizionemonomerica può essere più reattivo il monomero A o il mono-mero B. Si ottiene dunque la curva III in fig. 7, caratteriz-zata da un punto caratteristico, detto azeotropo, in cui le cate-ne polimeriche e la miscela monomerica da cui queste deri-vano hanno la stessa composizione. Analogo è il caso in cuile propagazioni dirette prevalgono su quelle incrociate, cioèrA�1 e rB�1.
Utilizzando il diagramma di Mayo e Lewis è possibileseguire anche in modo quantitativo il processo di deriva com-positiva precedentemente descritto. Si consideri un sistemadi copolimerizzazione il cui diagramma di Mayo e Lewis siaquello illustrato in fig. 8 e si supponga di introdurre inizial-mente nel reattore una miscela monomerica avente compo-sizione xA0. La prima quantità infinitesima di polimero pro-dotto ha la composizione FA0 indicata in f igura; essendoFA0�xA0, ciò implica che alla miscela monomerica viene sot-tratta una porzione più concentrata nel monomero A dellamiscela originaria. La conseguenza è che la frazione molaredi A, xA, nel reattore diminuisce, e con essa diminuisce la fra-zione molare dello stesso monomero nel polimero. Poichéperò la frazione molare di A nel polimero rimane superiorea quella nel monomero, entrambe continuano a diminuiredando luogo al processo di deriva compositiva. L’ampiezzadi tale variazione può essere apprezzata in modo qualitativoosservando che al termine del processo, quando i monomerihanno reagito completamente, la frazione molare media di Aglobalmente presente in tutte le catene polimeriche non puòche essere uguale a quella inizialmente presente nella fasemonomerica, cioè xA0. Dall’osservazione del diagramma difig. 8 è facile dedurre che affinché il valore medio di FA siauguale a xA0, avendo le prime catene composizione FA0, è neces-sario che le ultime catene prodotte abbiano una composizio-ne in A assai bassa.
Controllo della composizione del copolimero Nella maggior parte dei casi le caratteristiche applicative
di un copolimero richiedono un’elevata uniformità delle cate-ne polimeriche, particolarmente in termini della loro compo-sizione. Come visto precedentemente, tale risultato non è rea-lizzabile utilizzando un reattore discontinuo a causa del pro-cesso di deriva compositiva. Risulta pertanto necessario ricorrerea reattori semidiscontinui nei quali vengono introdotte duran-te il processo opportune quantità di monomeri, in modo damantenere costante nel tempo la composizione della miscelamonomerica reagente e conseguentemente delle catene pro-dotte durante il processo. Esistono due diverse strategie checonsentono di realizzare questo obiettivo.
Processo controllato dall’alimentazione. Questa modalitàoperativa si basa sul rallentamento del processo fino al puntoda consentire allo stesso di porsi, per la maggior parte deltempo, in condizioni di pseudostazionarietà, cosicché le velo-cità di consumo dei singoli monomeri siano uguali a quelle dialimentazione degli stessi, e dunque facilmente controllabili.Questa situazione viene raggiunta adottando un tempo di ali-mentazione tf molto elevato rispetto al tempo caratteristico diconsumo del monomero:
[71]
In questo modo il tempo caratteristico del processo coin-cide col tempo di alimentazione e il processo di consumo delmonomero, il cui tempo caratteristico risulta a questo puntoassai inferiore, è rispetto a esso in condizioni di pseudostazio-narietà.
Il processo controllato dall’alimentazione si realizza dun-que semplicemente alimentando una miscela monomerica cheabbia la stessa composizione di quella del polimero desidera-to, ma in modo sufficientemente lento da rispettare la condi-zione [71], senza necessariamente conoscere né la cinetica delprocesso né il relativo diagramma di Mayo e Lewis. Si ottie-ne così che per la maggior parte del processo vengono pro-dotte catene con la composizione desiderata, ma con lo svan-taggio di una diminuzione della produttività, in quanto il pro-cesso viene rallentato attraverso un’alimentazione molto lentarispetto alla velocità con cui i monomeri sarebbero in gradodi reagire; da qui la denominazione di processo starved (let-teralmente, affamato) usualmente utilizzata per questa moda-lità operativa.
In questi processi la concentrazione di monomero nel reat-tore si mantiene sostanzialmente costante per l’intero proces-so. Ciò può risultare particolarmente positivo in diverse situa-zioni, come per esempio quella discussa nel contesto dell’e-quazione [45], in cui si intende produrre una distribuzione dipesi molecolari molto stretta (per un omo- o un copolimero)nel caso in cui domini la terminazione bimolecolare per com-binazione.
Altre caratteristiche rilevanti di questi processi sono lafacilità del controllo termico, che deriva dalla bassa velocitàdi consumo dei monomeri, e il fatto che la conversione all’in-terno del reattore è sempre molto elevata. Ciò significa che laconcentrazione di polimero prevale decisamente su quella dimonomero e dunque tendono a prevalere le reazioni di tra-sferimento a polimero e le cinetiche controllate dall’effettoTrommsdorff.
Processo ad alimentazione forzata. Questa seconda mo-dalità operativa, usualmente denominata power feed, è piùambiziosa della precedente in quanto si propone non solo di
τ fpk R
>> 1
*
PROCESSI DI POLIMERIZZAZIONE
383VOLUME V / STRUMENTI
FA
FA0
xA0
xA
10 xA00
1
FA
FA0
xA0
xA
0
1
10 xA0
fig. 8. Rappresentazione schematica del processo di deriva compositiva in un reattore di copolimerizzazione chiuso, con composizione iniziale della miscela monomerica pari a xA0.

mantenere uniforme nel tempo la composizione delle cateneprodotte, ma anche di mantenere elevata la produttività del pro-cesso. La sua realizzazione richiede però maggiori conoscen-ze sulla cinetica della polimerizzazione, e in particolare sullarelazione di Mayo e Lewis che determina la composizionemonomerica necessaria per realizzare una desiderata compo-sizione istantanea della catena polimerica. La procedura richie-de quindi di porre nel reattore una certa quantità iniziale dimonomeri avente tale composizione e alimentare poi una cor-rente degli stessi in modo da mantenerla costante durante l’in-tero processo. In definitiva tale portata viene valutata in mododa alimentare la quantità necessaria a riequilibrare lo sbilan-ciamento di composizione dovuto alla diversa reattività deimonomeri. Attraverso opportuni bilanci materiali si ottiene laseguente relazione:
[72]
dove QA e QB sono le portate molari dei due monomeri chedevono essere alimentate, NA e NB sono le moli dei due mono-meri presenti all’interno del reattore a un certo istante e i ter-mini coinvolgenti le costanti pseudocinetiche di propagazionek*
pA e k*pB rappresentano le moli dei due monomeri consumate
dalle reazioni di propagazione per unità di tempo.Poiché l’equazione [72] rappresenta un legame tra le due
portate di alimentazione dei monomeri, rimane a questo puntodisponibile ancora un grado di libertà per l’ottimizzazione delprocesso. Questo può essere saturato per esempio richiedendoche il processo abbia la massima produttività, cioè la massimaquantità di polimero prodotto nell’unità di tempo. È possibiledimostrare che questo obiettivo viene raggiunto semplicementeintroducendo il monomero meno reattivo interamente all’ini-zio della reazione e alimentando quindi durante il processosolo il monomero più reattivo in modo da soddisfare la condi-zione [72], dove la portata del monomero meno reattivo vieneposta uguale a zero.
In questo caso insieme alla produttività aumenta la velo-cità di produzione del calore, e pertanto il reattore deve esse-re equipaggiato in modo opportuno per consentirne lo smalti-mento ed evitare pericolosi fenomeni di aumento incontrolla-to o runaway della temperatura.
Un’ultima osservazione riguarda l’applicazione pratica dellarelazione [72]. Questa richiede infatti che a ogni istante duran-te il processo siano noti tutti i termini che in essa compaiono.Ciò comporta, essendo coinvolti parametri cinetici, una buonaconoscenza della cinetica della reazione di polimerizzazione,ma anche di variabili di processo, quali per esempio le quan-tità dei due monomeri presenti nel reattore. È pertanto richie-sta l’utilizzazione in linea e in situ di opportuni sensori che con-sentano un monitoraggio affidabile di tali variabili. I sensoripiù spesso adottati a questo scopo sono basati sul bilancio ter-mico del reattore o su misure di altre grandezze indirettamen-te collegate alla conversione, come la densità o la velocità dipropagazione di ultrasuoni. È evidente che l’applicazione diquesta metododica operativa richiede uno sforzo maggiore delprocesso controllato dall’alimentazione, che deve dunque esse-re ricompensato dalla aumentata produttività del reattore.
6.4.6 Caratteristiche operative
Le reazioni di polimerizzazione radicalica possono essere con-dotte utilizzando processi con diverse caratteristiche operati-ve. Uno dei problemi più rilevanti si riferisce alla rimozione
del calore generato dalle reazioni di polimerizzazione. Questesono infatti reazioni fortemente esotermiche, paragonabili inquesto senso alle reazioni di combustione, e al crescere dellaconversione la miscela di reazione tende ad aumentare di visco-sità, con conseguente rallentamento dei processi di trasportotermico. Questa situazione rende difficile il controllo dellatemperatura del reattore causando indesiderate disuniformitànel prodotto finale o addirittura processi di aumento incon-trollato della temperatura con conseguenze che possono esse-re rilevanti anche sotto il profilo della sicurezza. In questi casiinfatti la temperatura aumenta in modo autoaccelerato in con-seguenza del fatto che un aumento di temperatura produce,attraverso una maggiore velocità di polimerizzazione, unaumento della velocità di produzione del calore, che a suavolta causa un ulteriore aumento di temperatura e così via. Aciò si aggiunga che al procedere della reazione la rimozionedel calore può essere rallentata dall’aumento di viscosità dellamiscela reagente, rendendo la temperatura del reattore anco-ra meno controllabile.
I processi di polimerizzazione si classificano in generalecome omogenei o eterogenei con riferimento alla miscela rea-gente. Questa si mantiene omogenea nei processi di polime-rizzazione in massa e in soluzione. Nel primo caso vengonointrodotti nel reattore esclusivamente il monomero e l’inizia-tore. Si ottiene dunque un prodotto a elevata purezza, partico-larmente apprezzabile in varie applicazioni. Il problema stanello smaltimento del calore di reazione, particolarmente dif-ficile quando, con l’aumentare della conversione, la viscositàdel sistema aumenta. Questi processi vengono comunque adot-tati per esempio nella produzione in continuo di vari tipi digomme, evitando di raggiungere nel reattore condizioni di con-versione completa e procedendo alla devolatilizzazione delmonomero che non ha reagito in opportune unità di separa-zione. Un altro esempio è la produzione di cere, basate su oli-gomeri dello stirene, che viene condotta a temperature moltoelevate con viscosità simili a quelle dei liquidi.
Per evitare l’aumento di viscosità della miscela reagente èpossibile utilizzare la polimerizzazione in soluzione. Questarichiede l’impiego di un solvente che disciolga sia il mono-mero sia il polimero, in modo da mantenere omogenea la poli-merizzazione per l’intero processo. Oltre alla difficoltà di iden-tificare un tale solvente, che sia soddisfacente in termini eco-nomici e normativi, questi processi richiedono elevati volumiper far circolare importanti quantità di solvente, il che non con-tribuisce alla loro produttività.
I processi eterogenei, in sospensione e in emulsione, sibasano sull’idea di fare avvenire la polimerizzazione in pic-cole particelle disperse in una matrice acquosa. Le particellesono caratterizzate da un elevatissimo rapporto superficie/volu-me che consente un rapidissimo trasporto del calore di rea-zione dal loro interno alla massa acquosa, da cui il calore vienepoi facilmente rimosso mediante elementi di raffreddamentotradizionali, poiché la viscosità della fase continua si mantie-ne ovviamente assai modesta. Il risultato è una dispersione diparticelle di polimero in acqua che richiede opportuni proces-si di coagulazione ed essiccamento da condursi a valle del reat-tore. Questi processi vengono realizzati partendo da una disper-sione di gocce di monomero in acqua, stabilizzate con mecca-nismi di tipo ionico o sterico da opportuni emulsionanti.All’interno di tali dispersioni viene quindi introdotto l’inizia-tore, che può essere solubile nella fase monomerica dispersao in quella acquosa, dando luogo rispettivamente al processodi polimerizzazione in sospensione o in emulsione.
N Q k RN N Q k RNB A pA A A B pB B−( ) = −( )* *
ASPETTI PROCESSISTICI
384 ENCICLOPEDIA DEGLI IDROCARBURI

Nel primo caso le gocce di monomero e di iniziatore si com-portano come dei piccoli reattori segregati, ben controllati ter-micamente, all’interno dei quali avviene una normale reazionedi polimerizzazione in massa. In questo caso si ottiene unadispersione di particelle di polimero finale che è molto similea quella iniziale di gocce di monomero, con dimensioni mediedell’ordine dei 100–1.000 mm. Il comportamento cinetico è per-tanto analogo a quello dei processi di polimerizzazione omo-genea, per i quali valgono le relazioni e le considerazioni svol-te precedentemente. Diversa è la situazione della polimerizza-zione in emulsione. In questo caso i radicali prodotti in faseacquosa devono nucleare le particelle di polimero che poi cre-scono durante il processo. Conseguentemente la dispersionefinale di particelle di polimero è assai diversa da quella inizia-le di gocce di monomero, con dimensioni medie che sono infe-riori di tre o quattro ordini di grandezza, cioè sono dell’ordinedi 100 nm. In particelle così piccole le reazioni di terminazio-ne bimolecolare diventano rapidissime, tanto da consentire allimite la presenza di non più di una catena radicalica per parti-cella. Ciò dà luogo al fenomeno della segregazione dei radica-li, che rende la cinetica della polimerizzazione in emulsione ele caratteristiche del polimero finale completamente diverse daquelle dei precedenti processi, come discusso di seguito.
6.4.7 Cinetica della polimerizzazione in emulsione
Il meccanismo cinetico alla base della polimerizzazione inemulsione è stato identificato da W.V. Smith e R.H. Ewart esuccessivamente approfondito e descritto in termini matema-tici da diversi ricercatori. Il processo viene diviso in tre fasifondamentali. Nella prima, detta di nucleazione, vengono for-mate le particelle di polimero. Esistono due tipi di nucleazio-ne possibili. La nucleazione micellare avviene quando l’e-mulsionante è inizialmente presente nella miscela reagente inconcentrazione superiore alla sua concentrazione critica micel-lare. In questo caso nel sistema sono presenti delle micelle diemulsionante di dimensioni assai ridotte, tipicamente dell’or-dine di alcuni nanometri. Rispetto alle gocce di monomero,dell’ordine di almeno 100 mm, queste hanno dunque un rap-porto superficie/volume superiore di almeno quattro ordini digrandezza. Conseguentemente, nonostante il monomero siapresente in quantità assai superiore rispetto all’emulsionantenella miscela di reazione, la superficie totale delle micelle è dialmeno due ordini di grandezza superiore a quella delle goccedi monomero. Ciò giustifica il fatto che la maggior parte deiradicali prodotti nella soluzione acquosa diffonde nelle micel-le, dove inizia la polimerizzazione utilizzando le molecole dimonomero solubilizzate all’interno delle micelle, che da que-sto momento costituiscono particelle di polimero. Un mecca-nismo alternativo, che è dominante per esempio nelle polime-rizzazioni condotte in assenza di emulsionante, è il meccani-smo di nucleazione omogenea. In questo caso sono le stessemolecole di polimero prodotte in acqua che si aggregano a for-mare micelle eventualmente stabilizzate dall’emulsionante pre-sente. È da notare che, avendo spesso i prodotti di decompo-sizione dell’iniziatore natura ionica o comunque idrofila, lestesse catene polimeriche possono comportarsi come moleco-le di emulsionante.
In questo processo, il numero di particelle Np cresce neltempo secondo una legge che deriva dal sovrapporsi di diversiprocessi chimico-fisici. Per esempio, nel caso di nucleazione
micellare a mano a mano che nuove particelle vengono nuclea-te, queste cominciano a crescere e per mantenersi stabili adsor-bono sulla loro superficie le molecole di emulsionante pre-senti in acqua. Ciò causa la dissoluzione delle micelle presentiper mantenere la concentrazione di emulsionante in soluzio-ne uguale alla concentrazione critica micellare e conseguen-temente il rallentamento della velocità di nucleazione. Que-st’ultima termina quando tutte le micelle vengono consumateo per dissoluzione o perché trasformate in particelle polime-riche. Un’analisi matematica semplificata di questi processiporta a una dipendenza del numero di particelle finale dalleconcentrazioni iniziali di emulsionante e iniziatore che non èlineare, caratterizzata da un esponente pari a 0,6 per il primoe 0,5 per il secondo.
Al termine della nucleazione, la concentrazione di emul-sionante in acqua Ew scende al di sotto della concentrazionecritica micellare (c.m.c.) e da questo momento le particelle increscita non possono più adsorbire quantità significative diemulsionante dalla fase acquosa. Ciò può rendere le stessemeno stabili dal punto di vista colloidale e portare a fenome-ni di coagulazione parziale. Attraverso la formazione di aggre-gati più grossi la superficie da stabilizzare infatti si riduce edunque i problemi di stabilità si riducono. Per evitare coagu-lazioni e mantenere la dispersione stabile, è opportuno aggiun-gere emulsionante durante la reazione. Una buona regola inquesto senso è l’introduzione di emulsionante in modo pro-porzionale alla crescita della superficie totale delle particel-le. È opportuno notare che aggiunte eccessive potrebbero por-tare alla formazione di nuove micelle e quindi alla produzio-ne di una nuova generazione di particelle; si potrebbe cosìavere una dispersione finale con distribuzione bimodale, ocomunque molto ampia, delle dimensioni delle particelle poli-meriche.
In questo contesto è opportuno notare che in alcune appli-cazioni è desiderabile produrre dispersioni con distribuzionimonomodali e molto strette delle dimensioni delle particelle.Ciò può essere ottenuto rendendo la fase di nucleazione moltobreve rispetto alla durata del processo, in modo che tutte leparticelle abbiano sostanzialmente lo stesso tempo a disposi-zione per crescere. Secondo la trattazione di Smith ed Ewart,in presenza di nucleazione micellare la durata del periodo dinucleazione è proporzionale alla potenza 0,6 della concentra-zione iniziale di emulsionante e inversamente proporzionalealla stessa potenza della concentrazione iniziale di iniziatore.
Prima di procedere alla descrizione delle successive duefasi del processo di polimerizzazione in emulsione è opportu-no considerare il fenomeno della segregazione dei radicali inparticella che, come indicato precedentemente, ne rappresen-ta l’elemento distintivo. Nel seguito è riportata una descrizio-ne semplificata di questo processo (per una discussione piùdettagliata, v. cap. 8.1).
Si consideri una particella di polimero che non contengainizialmente alcun radicale e all’interno della quale dunque nonavviene alcuna reazione. Si indichi con te l’intervallo di tempomedio necessario affinché un radicale proveniente dalla solu-zione acquosa entri in una particella. Come illustrato in fig. 9,dopo un tempo te la particella riceve un radicale, che essendosolo può dare luogo unicamente a reazioni di propagazione (sitrascurano al momento le reazioni di trasferimento), fino a che,dopo un nuovo intervallo te, entra nella particella un secondoradicale. A questo punto i due radicali oltre che alla propa-gazione possono dare luogo a una terminazione bimolecolare,il cui tempo caratteristico sia tt. Ne segue che i due radicali
PROCESSI DI POLIMERIZZAZIONE
385VOLUME V / STRUMENTI

propagano insieme per un tempo tt, dopo di che ha luogo laterminazione e, come indicato in fig. 9, la particella torna nellasituazione iniziale di assenza di radicali. Trattandosi evidente-mente di un processo periodico, si può valutare il valore mediodel numero di radicali per particella, n̄, che, facendo riferimentoa un periodo del grafico in figura, è dato da
[73]
A causa della dimensione molto ridotta delle particelle dipolimero, la cinetica di incontro dei due radicali è estrema-mente elevata; ciò si riflette in un valore del corrispondentetempo caratteristico molto ridotto, cosicché ttte, e la rela-zione precedente porta a
[74] n̄ �0,5
cioè mediamente in questo processo a ogni istante metà delleparticelle ha un radicale e cresce, mentre l’altra metà rimaneinerte.
La seconda fase del processo, detta di crescita, è caratte-rizzata da una velocità globale di consumo del monomero, Rp,che rimane sostanzialmente costante nel tempo. Tale andamentopuò essere giustificato analizzando il comportamento del mono-mero. Questo si trova inizialmente disperso in forma di grossegocce all’interno della fase acquosa che, come visto preceden-temente, rimangono sostanzialmente inerti e in minima partesolubilizzate in fase acquosa, che risulta infatti satura di mono-mero. Le particelle di polimero che si formano durante il pro-cesso contengono anch’esse monomero ed è in particolare que-sto monomero che partecipa alla reazione di polimerizzazione,essendo questo il luogo dove è preponderante la concentrazio-ne di radicali. Poiché le specie monomeriche sono in generalescarsamente solubili in acqua, in questo stadio del processo lamaggior parte del monomero è presente nelle gocce di mono-mero e nelle particelle di polimero, che viene rigonfiato in pro-porzioni dell’ordine del 50% in volume. Il monomero vieneconsumato solo in queste ultime e si rende dunque necessarioun continuo trasporto del monomero dalle gocce (che dunqueagiscono come un serbatoio) alle particelle, dove viene consu-mato, attraverso la fase acquosa. Poiché entrambe le fasi disper-se hanno rapporti superficie/volume estremamente elevati, ilfattore controllante del processo è la reazione di polimerizza-zione e dunque la concentrazione di monomero si mantienecostante sia in soluzione acquosa che in particella e uguale aicorrispondenti valori di saturazione. La velocità con cui il mono-mero viene trasportato all’interno di una particella può dunque
essere valutata attraverso la velocità di consumo del monome-ro all’interno della singola particella, Rpp, che è data da
[75]
dove Mp è la concentrazione di monomero in particella, Vp ilvolume di particella e il termine n̄�(VpN) rappresenta la con-centrazione di radicali in particella, dove N è il numero di Avo-gadro. È facile notare che, poiché il volume di particella si eli-mina nell’equazione [75], il termine Rpp tende a mantenersicostante in questo stadio del processo, implicando che le par-ticelle crescono con velocità sostanzialmente costante. Consi-derando che il numero di particelle, Np, in questo stadio è costan-te, se ne conclude che la velocità globale di polimerizzazioneRp�NpRpp è anch’essa costante.
Lo stadio di crescita del processo si estende fino al momen-to in cui tutto il monomero contenuto nelle gocce iniziali vieneconsumato e le gocce spariscono dunque dal sistema. A que-sto punto inizia il terzo e ultimo stadio, detto di consumo, duran-te il quale il monomero in particella continua a essere consu-mato dalla reazione di polimerizzazione e, poiché non può piùessere rimpiazzato, la sua concentrazione Mp diminuisce neltempo, al di sotto del valore di saturazione, Mp
sat, fino a esau-rimento della reazione.
Dall’esame della cinetica del processo di polimerizzazio-ne in emulsione emergono alcune caratteristiche che lo ren-dono particolarmente interessante ai fini applicativi, al di làdegli aspetti di controllo della temperatura precedentementemenzionati.
In un processo classico di polimerizzazione radicalica omo-genea (inclusa la polimerizzazione in sospensione) in cui domi-nano le terminazioni bimolecolari, si è visto in precedenza chela velocità complessiva Rp di consumo del monomero è data da
[76]
mentre la lunghezza media delle catene è data, secondo la[45], da
[77]
Confrontando tali relazioni si osserva che la velocità dipolimerizzazione e la lunghezza delle catene polimeriche sonolegate attraverso la concentrazione dei radicali. In particolareaumentando quest’ultima migliora la produttività del proces-so ma si producono catene più corte. Risulta pertanto impos-sibile produrre catene ad alto peso molecolare e avere con-temporaneamente un’elevata produttività.
Questo legame non è presente nella polimerizzazione inemulsione a causa della presenza del fenomeno della segrega-zione dei radicali. Come appare infatti dalla discussione fattanel contesto della fig. 9, nel caso di terminazioni bimolecola-ri molto veloci, la catena polimerica ha un tempo di vita mediosostanzialmente uguale a te, cioè il tempo che mediamente tra-scorre tra l’entrata di due radicali in una particella. La lun-ghezza media delle catene così prodotte può dunque esserevalutata dividendo tale termine per il tempo caratteristico diaddizione di un’unità monomerica, dato da tp�1�kpMp, otte-nendo così
[78]
dove il tempo caratteristico te è proporzionale alla concentra-zione dei radicali in fase acquosa e alla superficie della parti-cella polimerica. D’altro canto, utilizzando la relazione [75] si
n k MN p p e= τ
nk Mk RN
p
t
=2
R k MRp p=
R k M nV N
Vpp p pp A
p=
n e t
e t
=+
+τ τ
τ τ2
2
ASPETTI PROCESSISTICI
386 ENCICLOPEDIA DEGLI IDROCARBURI
1
0
2
tempo
num
ero
di r
adic
ali i
n pa
rtic
ella
tt
te
te
tt
te
te
fig. 9. Numero di radicali in una particella in funzione del tempo.

osserva che la velocità globale di consumo del monomero,Rp�NpRpp, è data da
[79]
Confrontando le relazioni [78] e [79] appare chiaro che ledue grandezze sono ora disaccoppiate: la polimerizzazione inemulsione consente infatti di ottenere pesi molecolari più altidei processi di polimerizzazione omogenei (inclusa la poli-merizzazione in sospensione) a parità di produttività.
Un’altra peculiarità della reazione in emulsione è che perla maggior parte del processo (esclusa la terza fase) la con-centrazione di monomero in particella si mantiene costante;dunque la reazione si svolge a concentrazione costante di mono-mero rendendo più facile il controllo dell’architettura dellecatene prodotte e quindi più uniforme il prodotto finale. Ciòappare evidente per esempio dalla relazione [78], che indicacome il peso molecolare istantaneo delle catene prodotte siaproporzionale alla concentrazione di monomero in particella.
Bibliografia generale
Biesenberger J.A., Sebastian D.H. (1983) Principles ofpolymerization engineering, New York, John Wiley.
Cooper W. (1976) Kinetics of polymerization initiated by Ziegler-Natta catalysts, in: Bamford C.H., Tipper C.F.H. (edited by)Comprehensive chemical kinetics, Amsterdam, Elsevier, 1968-1976, 18v.; v.XV.
Fitch R.M. (edited by) (1971) Polymer colloids. Proceedings of anAmerican chemical society symposium on polymer colloid held inChicago, 13-18 September 1970, New York, Plenum Press.
Flory P.J. (1953) Principles of polymer chemistry, Ithaca (NY), CornellUniversity Press.
Gilbert R.G. (1995) Emulsion polymerization. A mechanistic approach,London, Academic Press.
Gupta S.K., Kumar A. (1987) Reaction engineering of step growthpolymerization, New York, Plenum Press.
Hamielec A.E., Tobita H. (1992) Polymerization processes, in:Ulmann’s encyclopedia of industrial chemistry, Weinheim, VCH,1985-1993, 37v.; v.A21, 317-336.
Lovell P.A., El-Aasser M.S. (edited by) (1997) Emulsionpolymerization and emulsion polymers, New York, John Wiley.
Mayo F.R., Lewis F.M. (1944) Copolymerization. I: A basis forcomparing the behaviour of monomers in copolymerization. Thecopolymerization of styrene and methyl methecrylate, «Journal ofthe American Chemical Society», 66, 1594-1601.
Min K.W., Ray W.H. (1974) On the mathematical modeling of emulsionpolymerization, «Journal of Macromolecular Science. Part C.Review in Macromolecular Chemistry», 11, 177.
Ray W.H. (1977) Polymerization reaction engineering, in: LapidusL., Amudson N.R. (editors) Chemical reactor theory, EnglewoodCliffs (NJ), Prentice-Hall.
Ray W.H. (1983) Current problems in polymerization reactionengineering, «American Chemical Society Symposium Series»,226, 101.
Reichert K.H., Moritz H.U. (1989) Polymer reaction engineering,in: Comprehensive polymer science: the synthesis, characterization,reactions & applications of polymers, New York, Pergamon Press,7v.; v.III, 327.
Seymour R.B., Carraher C.E. (1988) Polymer chemistry, New York,Marcel Dekker.
Smith W.V., Ewart R.H. (1948) Kinetics of emulsion polymerization,«Journal of Chemical Physics», 16, 592-599.
Tirrel M. et al. (1987) Polymerization reactors, in: Carberry J.J.,Varma A. (editors) Chemical reaction and reactor engineering,New York, Marcel Dekker.
Trommsdorff E. (1954) Pearl polymerization, «MakromolekulareChemie», 13, 76-89.
Elenco dei simboli
A concentrazione del monomero AA� concentrazione totale di radicali di tipo AA�n concentrazione di catene contenenti globalmente n
unità monomeriche con l’attività radicalica su un’unità di tipo A
B concentrazione del monomero BB� concentrazione totale di radicali di tipo BB�n concentrazione di catene contenenti globalmente n
unità monomeriche con l’attività radicalica su un’unità di tipo B
C concentrazione del monomero CC� concentrazione totale di radicali di tipo CEw concentrazione di emulsionante in fase acquosaf efficienza dell’iniziatorefN(n) distribuzione istantanea numerale delle lunghezze
di catenafW(n) distribuzione istantanea ponderale delle lunghezze
di catenafN
c(n,t) distribuzione cumulata numerale delle lunghezze di catena
Fi frazione molare del monomero i nel polimeroI concentrazione di iniziatorekd costante cinetica di decomposizione dell’iniziatorekfm costante cinetica di trasferimento al monomerokfp costante cinetica di trasferimento al polimerokfs costante cinetica di trasferimento al trasferitore
di catenakp costante cinetica di propagazione kpij costante cinetica di propagazione del radicale i
col monomero jkt ktc�ktd
ktc costante cinetica di terminazione bimolecolare per combinazione
ktd costante cinetica di terminazione bimolecolare per disproporzionamento
ktcij,ktdij costante cinetica di terminazione del radicale icol radicale j
M concentrazione di monomeroMp concentrazione di monomero in particellan̄ numero medio di radicali in particellanN media numerale del numero di unità monomeriche
in catenanW media ponderale del numero di unità monomeriche
in catenaN numero di AvogadroN(r) distribuzione numerale delle catene di lunghezza rNi moli di monomero iNp numero di particelle per unità di volumepi frazione molare dei radicali di tipo iP concentrazione totale di catene mortePn concentrazione di catene morte contenenti n unità
monomericheQi portata molare del monomero iri rapporto di reattività relativo al radicale di tipo i
R k M nN
Np p pA
p=
PROCESSI DI POLIMERIZZAZIONE
387VOLUME V / STRUMENTI

R concentrazione totale di radicali R costante dei gas idealiRn concentrazione di radicali contenenti n unità
monomeriche Rp velocità globale di polimerizzazioneRpi velocità globale di consumo del monomero iRpp velocità globale di consumo del monomero in una
particellaRI velocità di produzione di radicaliS concentrazione del trasferitore di catenat tempoT temperatura Vp volume della particellaW(r) distribuzione ponderale delle catene
di lunghezza rXi frazione molare del monomero i nella miscela
reagenteXA conversione del monomero A
Lettere grechea b�gb parametro cinetico adimensionale
tp12
ttc
g parametro cinetico adimensionale tp12
ttd�
tp12
tfm�
tp12
tfss polidispersità nW �nN
te tempo caratteristico di entrata di un radicale inparticella
tf tempo caratteristico dell’alimentazione
tfm tempo caratteristico della reazione di trasferimentoa monomero
tfs tempo caratteristico della reazione di trasferimentoal trasferitore di catena
tp tempo caratteristico della reazione di propagazionetI tempo caratteristico della reazione di
decomposizione dell’iniziatorett ttc�ttd
ttc tempo caratteristico della reazione di terminazioneper combinazione
ttd tempo caratteristico della reazione di terminazioneper disproporzionamento
tM tempo caratteristico del processo di polimerizzazione
Apicisat condizioni di saturazione* costante pseudocinetica
PediciA monomero AB monomero BC monomero C0 valore iniziale
Massimo Morbidelli
Institut für Chemie- und BioingenieurwissenschaftenEidgenössische Technische Hochschule-Zürich
Zurigo, Svizzera
ASPETTI PROCESSISTICI
388 ENCICLOPEDIA DEGLI IDROCARBURI


















