
6.2 Processi di separazione - Treccani, il portale del sapere · La distillazione è il più...
32
319 VOLUME V / STRUMENTI 6.2.1 Caratteristiche dei processi di separazione La separazione di soluzioni e miscele nei loro singoli com- ponenti costituisce un’operazione di grande importanza per l’industria chimica, petrolchimica e petrolifera. Quasi tutti i processi chimici richiedono una purificazione preliminare delle materie prime o la separazione dei prodotti primari da quelli secondari. Queste operazioni si contrappongono alla tendenza delle sostanze a mescolarsi intimamente e sponta- neamente tra loro, che come è noto costituisce una tipica conseguenza del secondo principio della termodinamica. Per esempio, il sale mescolato con acqua si scioglie per dare una soluzione omogenea e la separazione dei componenti pre- senti nella miscela richiede l’impiego di energia. In questo caso la separazione può essere effettuata in uno dei modi seguenti: • fornendo calore alla soluzione per far evaporare l’acqua, condensandola successivamente a più bassa temperatura; • raffreddando la soluzione per separare l’acqua sotto forma di ghiaccio; • sfruttando le proprietà perselettive di una membrana che viene attraversata dall’acqua in modo preferenziale rispet- to al sale. I processi di separazione sono cruciali negli impianti petro- liferi. Il greggio infatti contiene un numero molto elevato di idrocarburi, che vanno dai gas leggeri alle frazioni pesanti, difficilmente distillabili anche sotto vuoto. In particolare le classi più importanti sono costituite dalle paraffine, dalle cicloparaffine (nafteni) e dai composti aromatici in varie pro- porzioni. Nelle raffinerie vengono separate le varie frazioni mediante distillazione, che successivamente sono ulterior- mente trattate per fornire i diversi prodotti di specifico inte- resse. Solitamente il greggio subisce dapprima un lavaggio con acqua per rimuovere i sali e le eventuali particelle sospe- se, quindi viene vaporizzato in un forno che lo porta a una temperatura di circa 400 °C. I vapori di greggio sono inviati in una colonna di distillazione, o torre di raffinazione, dove viene realizzata la separazione delle diverse frazioni di idro- carburi: nel punto più basso della colonna condensano oli combustibili, lubrificanti, paraffine, cere e bitumi; tra 350 °C e 250 °C condensa il gasolio, utilizzato come combustibi- le per i motori diesel e per il riscaldamento domestico; tra 250 °C e 160 °C condensa il cherosene, un combustibile oleo- so usato per impianti di riscaldamento e come propellente per aerei a reazione; tra 160 °C e 70 °C condensa la naphtha, usata come combustibile e come materia prima per pestici- di, fertilizzanti e materiali plastici. Le benzine, principal- mente usate come carburante per automobili e aerei, conden- sano tra 70 °C e 20 °C. A 20 °C rimangono i prodotti gasso- si quali il metano, l’etano, il propano e il butano. In particolare, butano e propano formano il carburante denominato GPL (Gas di Petrolio Liquefatto). L’esempio precedente mostra come un processo di separa- zione consente di trasformare una miscela di sostanze in due o più prodotti di differente composizione. L’alimentazione di un processo separativo è costituita da una o più correnti, men- tre dal dispositivo di separazione escono le correnti dei pro- dotti a diversa composizione. La separazione è provocata da un agente di separazione che può essere costituito da un’altra corrente di materia, o da un flusso di energia, o da entrambi. Spesso i processi di separazione danno luogo alla formazione di un’ulteriore fase, diversa rispetto a quella di alimentazione. Per esempio, alimentando una corrente liquida, i prodotti pos- sono essere costituiti da due correnti formate da un liquido e da un vapore. Tenendo conto di quanto precede, è possibile formulare una classificazione generale dei processi di separazione più utilizzati nell’industria. Essa è riportata in tab. 1, dove sono riassunte le loro caratteristiche essenziali. È opportuno caratterizzare i processi di separazione median- te un fattore di separazione, definito come segue: [1] dove x i indica la frazione molare del componente i e x j indica la frazione molare del componente j, mentre gli indici 1 e 2 indicano le due correnti dei prodotti della separazione. Per- tanto a s ij rappresenta il rapporto tra le frazioni molari dei due componenti i e j nelle due correnti 1 e 2. Quindi se a s ij 1 il processo non permette di ottenere alcuna separazione tra i com- ponenti i e j. Se a s ij 1, il componente i tende a concentrarsi nella corrente 1, mentre se a s ij 1, tale comportamento si ma- nifesta per il componente j. Convenzionalmente si scelgono i due componenti in modo tale che a s ij risulti sempre maggio- re dell’unità. α ij s i j i j x x x x = 1 1 2 2 6.2 Processi di separazione
Transcript of 6.2 Processi di separazione - Treccani, il portale del sapere · La distillazione è il più...

319VOLUME V / STRUMENTI
6.2.1 Caratteristiche dei processi di separazione
La separazione di soluzioni e miscele nei loro singoli com-ponenti costituisce un’operazione di grande importanza perl’industria chimica, petrolchimica e petrolifera. Quasi tuttii processi chimici richiedono una purificazione preliminaredelle materie prime o la separazione dei prodotti primari daquelli secondari. Queste operazioni si contrappongono allatendenza delle sostanze a mescolarsi intimamente e sponta-neamente tra loro, che come è noto costituisce una tipicaconseguenza del secondo principio della termodinamica. Peresempio, il sale mescolato con acqua si scioglie per dare unasoluzione omogenea e la separazione dei componenti pre-senti nella miscela richiede l’impiego di energia. In questocaso la separazione può essere effettuata in uno dei modiseguenti: • fornendo calore alla soluzione per far evaporare l’acqua,
condensandola successivamente a più bassa temperatura; • raffreddando la soluzione per separare l’acqua sotto forma
di ghiaccio; • sfruttando le proprietà perselettive di una membrana che
viene attraversata dall’acqua in modo preferenziale rispet-to al sale.I processi di separazione sono cruciali negli impianti petro-
liferi. Il greggio infatti contiene un numero molto elevato diidrocarburi, che vanno dai gas leggeri alle frazioni pesanti,difficilmente distillabili anche sotto vuoto. In particolare leclassi più importanti sono costituite dalle paraffine, dallecicloparaffine (nafteni) e dai composti aromatici in varie pro-porzioni. Nelle raffinerie vengono separate le varie frazionimediante distillazione, che successivamente sono ulterior-mente trattate per fornire i diversi prodotti di specifico inte-resse. Solitamente il greggio subisce dapprima un lavaggiocon acqua per rimuovere i sali e le eventuali particelle sospe-se, quindi viene vaporizzato in un forno che lo porta a unatemperatura di circa 400 °C. I vapori di greggio sono inviatiin una colonna di distillazione, o torre di raffinazione, doveviene realizzata la separazione delle diverse frazioni di idro-carburi: nel punto più basso della colonna condensano olicombustibili, lubrificanti, paraffine, cere e bitumi; tra 350 °Ce 250 °C condensa il gasolio, utilizzato come combustibi-le per i motori diesel e per il riscaldamento domestico; tra
250 °C e 160 °C condensa il cherosene, un combustibile oleo-so usato per impianti di riscaldamento e come propellente peraerei a reazione; tra 160 °C e 70 °C condensa la naphtha,usata come combustibile e come materia prima per pestici-di, fertilizzanti e materiali plastici. Le benzine, principal-mente usate come carburante per automobili e aerei, conden-sano tra 70 °C e 20 °C. A 20 °C rimangono i prodotti gasso-si quali il metano, l’etano, il propano e il butano. In particolare,butano e propano formano il carburante denominato GPL(Gas di Petrolio Liquefatto).
L’esempio precedente mostra come un processo di separa-zione consente di trasformare una miscela di sostanze in dueo più prodotti di differente composizione. L’alimentazione diun processo separativo è costituita da una o più correnti, men-tre dal dispositivo di separazione escono le correnti dei pro-dotti a diversa composizione. La separazione è provocata daun agente di separazione che può essere costituito da un’altracorrente di materia, o da un flusso di energia, o da entrambi.Spesso i processi di separazione danno luogo alla formazionedi un’ulteriore fase, diversa rispetto a quella di alimentazione.Per esempio, alimentando una corrente liquida, i prodotti pos-sono essere costituiti da due correnti formate da un liquido eda un vapore.
Tenendo conto di quanto precede, è possibile formulareuna classificazione generale dei processi di separazione piùutilizzati nell’industria. Essa è riportata in tab. 1, dove sonoriassunte le loro caratteristiche essenziali.
È opportuno caratterizzare i processi di separazione median-te un fattore di separazione, definito come segue:
[1]
dove xi indica la frazione molare del componente i e xj indicala frazione molare del componente j, mentre gli indici 1 e 2indicano le due correnti dei prodotti della separazione. Per-tanto as
ij rappresenta il rapporto tra le frazioni molari dei duecomponenti i e j nelle due correnti 1 e 2. Quindi se as
ij�1 ilprocesso non permette di ottenere alcuna separazione tra i com-ponenti i e j. Se as
ij�1, il componente i tende a concentrarsinella corrente 1, mentre se as
ij�1, tale comportamento si ma-nifesta per il componente j. Convenzionalmente si scelgonoi due componenti in modo tale che as
ij risulti sempre maggio-re dell’unità.
αijs i j
i j
x xx x= 1 1
2 2
6.2
Processi di separazione
6.2.2 Bilanci di materia e di energia delle apparecchiaturedi separazione
Un impianto continuo di separazione può essere considera-to come un sistema termodinamico aperto agli scambi dimateria e di energia. A un dispositivo di separazione è pos-sibile associare flussi di materia che corrispondono alle cor-renti di alimentazione e ai prodotti di separazione, oltre cheflussi di energia necessari affinché possa aver luogo la sepa-razione. In condizioni normali gli apparati continui funzio-nano in regime stazionario, per cui i valori dei parametriintensivi del sistema (pressione, temperatura e concentra-zioni dei diversi componenti) non dipendono dal tempo, masono diversi nei diversi punti del sistema stesso. I loro gra-dienti, infatti, determinano le velocità dei processi con cuiavvengono i trasferimenti di materia e di energia nelle diver-se regioni del sistema.
Bilanci di materiaA un dispositivo di separazione si deve applicare il princi-
pio di conservazione della materia, che in termini generali (con-dizioni non stazionarie) si può esprimere come segue:
Il bilancio può essere applicato sull’intera apparecchiatu-ra o su qualunque sua porzione scelta arbitrariamente. Nelseguito si assume che all’interno delle apparecchiature di sepa-razione non avvengano reazioni chimiche e quindi vengonoposti uguali a zero i termini di generazione e consumo di mate-ria. Il bilancio assume quindi la forma semplificata: [accumu-lo]�[ingresso]�[uscita].
Per poterlo formulare in termini quantitativi, si indicanocon mi la massa del componente i contenuta nel sistema e conFi
(e) e Fi(u) le portate in massa dello stesso componente, rispet-
tivamente entranti e uscenti nell’apparecchiatura in esame. Ilbilancio materiale si può allora scrivere
[2]
Le sommatorie al secondo membro si intendono eseguitesu tutte le correnti entranti e uscenti. L’equazione precedenteesprime il bilancio di materia sul componente i. Sommando leequazioni di bilancio relative ai diversi componenti si ottienel’equazione di bilancio totale di materia:
[3]dmdt F Fe
e
u
u
= −( ) ( )∑ ∑
dmdt F Fi
ie
iu
ue
= −( ) ( )∑∑
consumo a− lll'interno del sistema[ ]
genera+ zzione all interno del sistema’[ ] −
tra− ssporto verso l esterno attraversola superf
’iicie che delimita il sistema
+
= ttrasporto verso l interno attraversola sup
’eerficie che delimita il sistema
−
accumulo di materiaentro il sistema
=
ASPETTI PROCESSISTICI
320 ENCICLOPEDIA DEGLI IDROCARBURI
tab. 1. Classificazione dei processi di separazione
Tipo di processo Alimentazione Agente di separazione Prodotti Principio di separazione
Evaporazione Liquido Calore Liquido e vapore Differenza in volatilità
Distillazione Liquido Calore Liquido e vapore Differenza in volatilità
Assorbimento Gas Liquido non volatile Liquido e gas Solubilità preferenziale
Estrazione Liquido Liquido immiscibile Due liquidi Differenti solubilità
Cristallizzazione LiquidoCalore (riscaldamento
o raffreddamento)Liquido e solido
Differenza nella temperaturadi cristallizzazione
Adsorbimento Gas o liquido Solido adsorbente Fluido e solidoDifferenza nelle caratteristiche
di adsorbimento
Scambio ionico Liquido Resina solida Liquido e solido Equilibrio di adsorbimento
Estrazione solido-liquido Solido Liquido Liquido e solido Diffusione e osmosi
Essiccamento Solido Calore Solido e vapore Differenza in volatilità
Sedimentazionee centrifugazione
Slurry, dispersioni Forza gravitazionale Solido e liquido Differenza in densità
Filtrazione Sospensione Filtro Solido e liquido Differenza in dimensioni
Processi a membrana Gas o liquido Membrana Gas o liquidoDifferenza in dimensionio differenza in solubilità
nella membrana
Flottazione Sospensione Agenti collettori Solido e liquido Tensione superficiale

In regime stazionario mi e m non variano con il tempo, equindi la [2] e la [3] diventano:
[4]
In modo del tutto analogo si possono scrivere le equazionidi bilancio molare riferite alle specie i e alle moli totali. Se siindica con ni il numero di moli del componente i contenute nelsistema e con Fi la portata molare del componente si ottiene:
[5]
che in regime stazionario diventa:
[6]
Bilanci di energiaUna caratteristica delle apparecchiature chimiche è la pre-
senza di movimenti di correnti fluide nelle quali possono avereluogo trasformazioni. Per poter scrivere le equazioni di bilan-cio di energia per le apparecchiature di separazione è quindinecessario combinare meccanica dei fluidi e termodinamica.Si consideri, per esempio, un sistema in cui un fluido scorrecon continuità in un condotto fra due sezioni 1 e 2, la cui quotarispetto a un piano di riferimento è diversa. Una pompa forni-sce un lavoro W e uno scambiatore di calore fornisce (o sot-trae) una quantità di calore Q. Riferendosi alla massa unitaria,l’energia entrante o uscente dalle due sezioni considerate è datadalla somma dei termini seguenti: energia interna riferita all’u-nità di massa U; energia potenziale relativa al piano di riferi-mento (riferita all’unità di massa essa è espressa come F�gz,dove g è l’accelerazione di gravità e z è la quota della sezionevalutata lungo un asse verticale); energia cinetica, che riferitaall’unità di massa è K�(1/2)u2, dove u è la velocità media incorrispondenza della sezione considerata.
La variazione di energia subita dalla massa dm nel pas-saggio dalla sezione 1 alla sezione 2 è data da:
[7]
ove l’operatore D indica la differenza fra i valori corrispon-denti alle due sezioni.
In base al principio di conservazione dell’energia, tale dif-ferenza, in condizioni stazionarie, dovrà uguagliare la sommadell’energia che il sistema riceve dall’ambiente e quindi:
[8]
Nel terzo membro della [8], dW è scisso nella somma didue termini. P1 e P2 rappresentano le pressioni presenti in cor-rispondenza delle sezioni considerate, mentre V1 e V2 sono ivolumi per massa unitaria. (P1V1�P2V2)dm rappresenta quindiil lavoro associato alla variazione di pressione che il fluido
subisce passando dalla sezione 1 alla sezione 2; dWs è inveceil lavoro eseguito sul sistema mediante un dispositivo mecca-nico, oppure sottratto mediante una turbina. La [8] si può quin-di scrivere nella forma:
[9]
Ricordando la definizione della funzione entalpia H�U�PVe dividendo tutti i termini dell’equazione per dt si ottiene:
[10]
dove m� è la portata in massa, mentre W�s e Q� rappresentano
rispettivamente le quantità di energia meccanica e termica for-nite al sistema nell’unità di tempo. Dividendo quindi la [10]per m� si ottiene:
[11]
dove Ws e Q sono rispettivamente il lavoro e il calore scam-biato per massa unitaria di fluido fluente.
La [11] può essere estesa a sistemi con più correnti entran-ti e più correnti uscenti, e in questo caso si deve effettuare ladifferenza tra la somma dei valori delle grandezze relative atutte le correnti uscenti e la somma dei valori delle grandezzerelative a tutte le correnti entranti. Spesso nell’analisi delleapparecchiature chimiche i termini di energia potenziale e cine-tica vengono trascurati e la [11] diventa semplicemente:
[12]
Espresso in questa forma, il bilancio energetico viene chia-mato bilancio entalpico o termico.
6.2.3 Distillazione
La distillazione è il più importante e diffuso metodo di sepa-razione dei componenti presenti in una miscela liquida ed èbasato sulla loro distribuzione tra la fase liquida e la fase vapo-re, quando la miscela viene portata all’ebollizione. La fattibi-lità e l’attrattiva economica di un processo di distillazione dipen-dono da molti fattori, tra i quali è importante menzionare lecaratteristiche favorevoli dell’equilibrio liquido-vapore, la com-posizione dell’alimentazione, il numero dei componenti daseparare, la purezza richiesta, la pressione assoluta necessariaper poter eseguire l’operazione, la stabilità al calore e la cor-rosività delle miscele.
Il primo di questi fattori si manifesta attraverso i valoridella volatilità relativa dei componenti ed è quello predomi-nante, perché influisce significativamente sull’energia e sulledimensioni dell’apparecchiatura richiesta per ottenere il desi-derato grado di purezza. Peraltro, la volatilità relativa di duecomponenti può essere modificata mediante l’aggiunta di unterzo componente (in questo caso si parla di distillazione estrat-tiva o azeotropica), oppure diminuendo la pressione assoluta.
Distillazione continua in uno stadio (flash)Come primo esempio di processo di separazione sarà con-
siderato un processo, detto normalmente flash, in cui una misce-la liquida viene parzialmente vaporizzata in un unico stadio.Un suo tipico schema viene illustrato in fig. 1. L’alimentazio-ne liquida viene riscaldata, per esempio attraversando uno scam-biatore tubolare, per cui quando la pressione viene ridotta siforma adiabaticamente vapore a spese del contenuto termicodel liquido. La miscela viene quindi inviata in un recipientedove ha luogo la separazione tra le due fasi, liquido e vapore.
∆ � � �H W Qs= +
∆ ∆ ∆� � � � �H K W Qs+ + = +Φ
∆ ∆ ∆� � � � � �H K m W Qs+ +( ) = +Φ
∆ ∆ ∆ ∆� � � �U K PV dm W Qs+ + + ( ) = +Φ δ δ
� �Q PV PV dm= + −( ) +δ δ1 1 2 2 WWs
∆ ∆ ∆� � �U K dm Q W+ +( ) = + =Φ δ δ
�U= ∆ ++ +( )∆ ∆� �Φ K dm
� �U U g z z u u dm2 1 2 1 2
2
1
21
2−( ) + −( ) + −( )
=
� �
� �
F F
F F
ie
iu
ue
e
e
u
u
( ) ( )
( ) ( )
=
=
∑∑∑ ∑
dndt F F
dndt F F
iie
iu
ue
e
e
u
u
= −
= −
( ) ( )
( ) ( )
∑∑
∑ ∑
� �
� �
F F
F F
ie
iu
ue
e
e
u
u
( ) ( )
( ) ( )
=
=
∑∑∑ ∑
PROCESSI DI SEPARAZIONE
321VOLUME V / STRUMENTI

In un modello ideale del processo si assume che le due fasipresenti nel separatore si trovino in condizioni di equilibrio ter-modinamico. In questo caso il flash rappresenta un esempio diseparazione per distillazione che ha luogo in un unico stadioideale ed è possibile definire un parametro di ripartizione Ki ,valutabile mediante la termodinamica:
[13]
dove xi,1 e xi,2 sono le frazioni molari del componente rispetti-vamente nelle fasi 1 e 2. Se, come nel caso presente, le due fasisono un vapore e un liquido, il parametro Ki viene chiamatorapporto di vaporizzazione:
[14]
dove yi e xi sono le frazioni molari del componente i rispetti-vamente nelle fasi vapore e liquido. Nel caso di miscele di idro-carburi i valori dei parametri Ki sono spesso riportati sottoforma di nomogrammi.
Il fattore di separazione definito nella [1] diventa:
[15]
anche detto volatilità relativa del componente i rispetto al com-ponente j, e dipende soltanto dalle caratteristiche termodina-miche della miscela da separare.
Tornando al problema del flash, l’equazione di bilanciomateriale totale dell’apparecchiatura di fig. 1 si può scrivere:
[16]
dove F è la portata molare (moli/tempo) dell’alimentazione, Vè quella del vapore e L quella del liquido. Il bilancio materia-le per il generico componente i è invece:
[17]
dove zi rappresenta la frazione molare di i nell’alimentazione,yi la frazione molare di i in fase vapore e xi quella in fase liqui-da. Combinando la [16] e la [17] si ottiene:
[18]
da cui:
[19]
Sostituendo la [14] nella [17] si ottiene:
[20]
che risolta rispetto a xi dà:
[21]
Le yi si possono invece ottenere combinando quest’ultimaequazione con la [14]:
[22]
Nel caso in cui le Ki dipendano solo da T e P, operando apressione assegnata, è possibile calcolare il valore della tem-peratura presente nell’apparecchiatura, ricordando che la sommadelle frazioni molari dei diversi componenti in entrambe le fasideve essere uguale a 1:
[23]
Sostituendo la [21] nella prima delle [23] si ottiene:
[24]
La temperatura quindi deve avere un valore tale da soddi-sfare l’equazione precedente.
La quantità di calore necessaria per il processo in questio-ne può essere infine calcolata mediante un bilancio energeti-co, espresso dalla [12], ponendo Ws uguale a zero perché nelsistema non sono presenti organi meccanici. Si ha quindi:
[25]
dove H è l’entalpia molare del vapore, h l’entalpia molare delliquido, hF l’entalpia molare dell’alimentazione e Q il caloreda fornire nell’unità di tempo.
Distillazione continua a più stadiSe si effettua la distillazione in un unico stadio, o flash, di
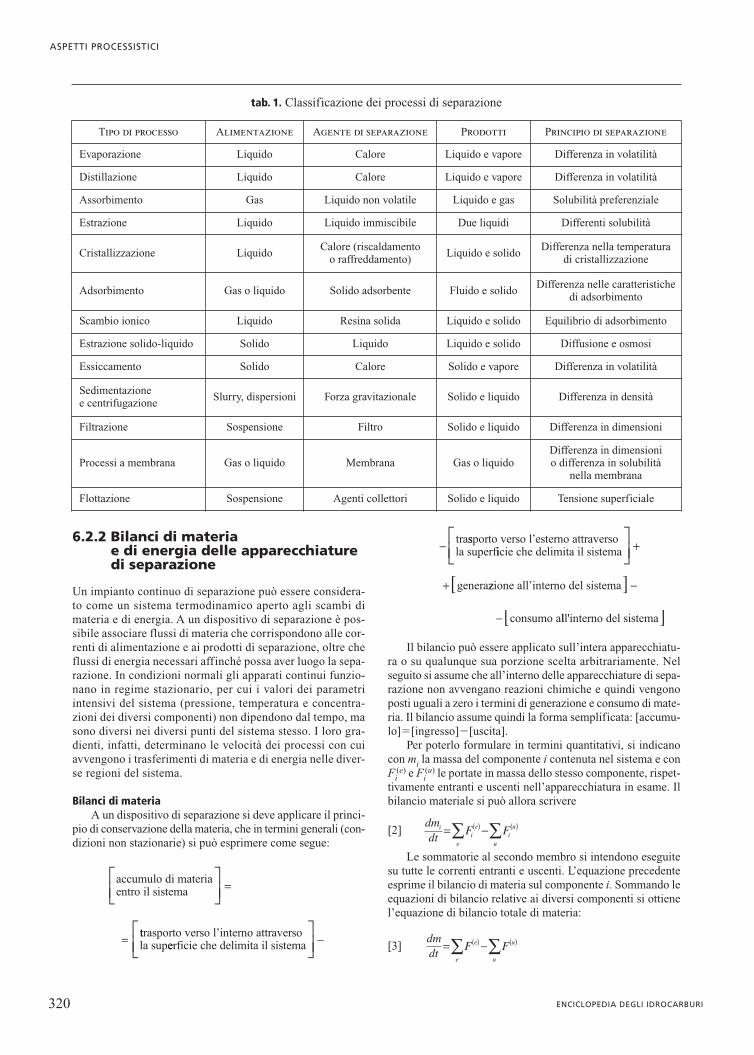
una miscela di due componenti, si ottengono come prodotti duefasi: un liquido ricco del componente meno volatile e un vapo-re ricco di quello più volatile, ma con un grado di separazionein generale piuttosto basso. La purezza del prodotto più volati-le può essere aumentata se si condensa parte del vapore pro-dotto, che viene successivamente vaporizzato realizzando unprocesso in due stadi. Se il procedimento viene ripetuto più volte,si può arrivare a ottenere un prodotto di testa (il più volatile)con un elevato grado di purezza. La stessa operazione può esse-re condotta sul liquido del primo processo di evaporazione, effet-tuando in stadi successivi diverse evaporazioni. Questo proces-so fornisce però quantità esigue di prodotto, poiché gli stadi suc-cessivi di evaporazione impoveriscono con continuità la correnteliquida che lascia il primo evaporatore. Analogamente, gli stadisuccessivi di condensazione impoveriscono con continuità lacorrente di vapore che lascia il primo evaporatore. Questo incon-veniente può essere evitato operando secondo lo schema indi-cato in fig. 2 in cui, nella parte superiore all’alimentazione, illiquido prodotto in uno stadio generico m alimenta lo stadio chelo precede (m�1), così come nella porzione inferiore all’ali-mentazione il vapore prodotto in un generico stadio n alimentalo stadio (n�1). Tale schema illustra l’insieme delle operazio-ni che hanno luogo in un processo di distillazione continua, cheviene infatti attuata attraverso una serie di stadi successivi, ognu-no dei quali può essere assimilato a un flash.
Nella pratica industriale l’operazione globale viene effet-tuata in una colonna di distillazione, costituita da una unitàcilindrica verticale in cui sono presenti più stadi e strutturatasecondo lo schema rappresentato nella fig. 3. In corrisponden-za di ciascuno stadio ha luogo un contatto intimo tra il vapore
� � � �Q VH Lh FhF= + −
FV
zK T L V
i
ii ( ) + =∑ 1
x
y
ii
ii
=
=
∑∑
1
1
y FV
K zK L Vi
i i
i
=+
x FV
zK L V
LV
zK L Vi
i
i
i
i
=+
= +
+
1
Fz x VK Li i i= +( )
LV
z yx zi i
i i= −
−
Vz Lz Vy Lxi i i i+ = +
Fz Vy Lxi i i= +
F L V= +
αiji j
i j
y xx y=
Kyxii
i
=
Kxxii
i
= ,
,
1
2
ASPETTI PROCESSISTICI
322 ENCICLOPEDIA DEGLI IDROCARBURI
vapore
liquido
alimentazione
riscaldatore
fig. 1. Schema di processo di distillazione continua a un solo stadio (flash).
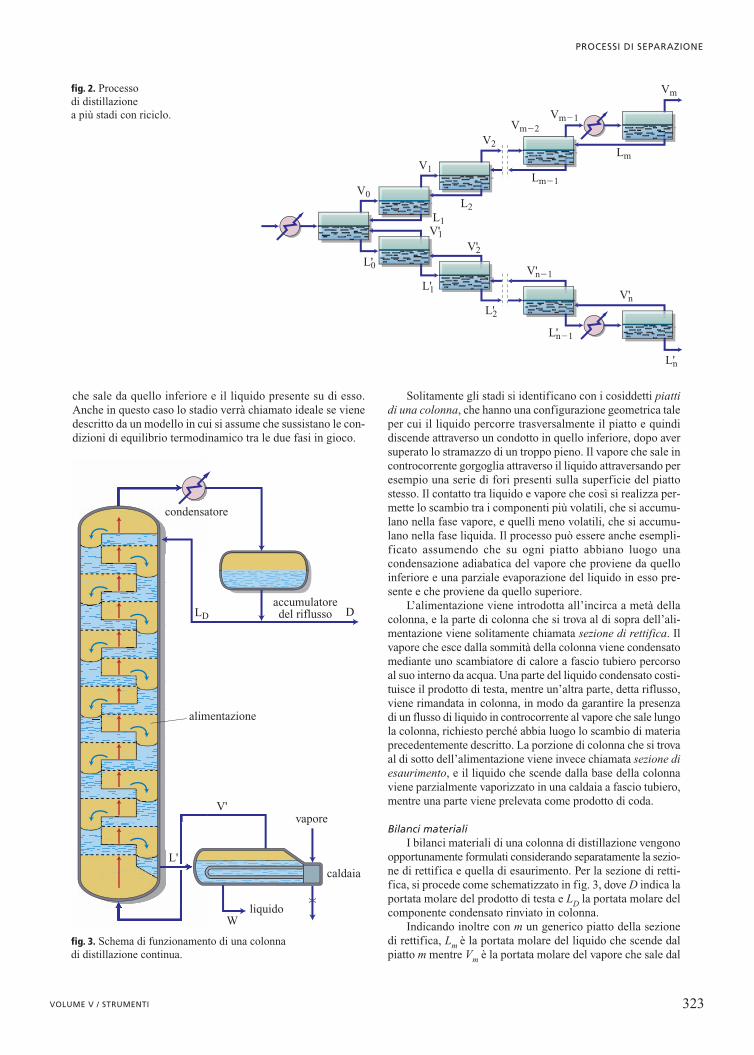
che sale da quello inferiore e il liquido presente su di esso.Anche in questo caso lo stadio verrà chiamato ideale se vienedescritto da un modello in cui si assume che sussistano le con-dizioni di equilibrio termodinamico tra le due fasi in gioco.
Solitamente gli stadi si identificano con i cosiddetti piattidi una colonna, che hanno una configurazione geometrica taleper cui il liquido percorre trasversalmente il piatto e quindidiscende attraverso un condotto in quello inferiore, dopo aversuperato lo stramazzo di un troppo pieno. Il vapore che sale incontrocorrente gorgoglia attraverso il liquido attraversando peresempio una serie di fori presenti sulla superficie del piattostesso. Il contatto tra liquido e vapore che così si realizza per-mette lo scambio tra i componenti più volatili, che si accumu-lano nella fase vapore, e quelli meno volatili, che si accumu-lano nella fase liquida. Il processo può essere anche esempli-ficato assumendo che su ogni piatto abbiano luogo unacondensazione adiabatica del vapore che proviene da quelloinferiore e una parziale evaporazione del liquido in esso pre-sente e che proviene da quello superiore.
L’alimentazione viene introdotta all’incirca a metà dellacolonna, e la parte di colonna che si trova al di sopra dell’ali-mentazione viene solitamente chiamata sezione di rettifica. Ilvapore che esce dalla sommità della colonna viene condensatomediante uno scambiatore di calore a fascio tubiero percorsoal suo interno da acqua. Una parte del liquido condensato costi-tuisce il prodotto di testa, mentre un’altra parte, detta riflusso,viene rimandata in colonna, in modo da garantire la presenzadi un flusso di liquido in controcorrente al vapore che sale lungola colonna, richiesto perché abbia luogo lo scambio di materiaprecedentemente descritto. La porzione di colonna che si trovaal di sotto dell’alimentazione viene invece chiamata sezione diesaurimento, e il liquido che scende dalla base della colonnaviene parzialmente vaporizzato in una caldaia a fascio tubiero,mentre una parte viene prelevata come prodotto di coda.
Bilanci materialiI bilanci materiali di una colonna di distillazione vengono
opportunamente formulati considerando separatamente la sezio-ne di rettifica e quella di esaurimento. Per la sezione di retti-fica, si procede come schematizzato in fig. 3, dove D indica laportata molare del prodotto di testa e LD la portata molare delcomponente condensato rinviato in colonna.
Indicando inoltre con m un generico piatto della sezionedi rettifica, Lm è la portata molare del liquido che scende dalpiatto m mentre Vm è la portata molare del vapore che sale dal
PROCESSI DI SEPARAZIONE
323VOLUME V / STRUMENTI
V0
V1
L1V'1
L'0
L'1
V'2
L'2
V'n�1
L'n�1
V'n
L'n
V2
L2
Vm�2
Lm�1
Vm�1
Vm
Lm
fig. 2. Processo di distillazione a più stadi con riciclo.
condensatore
accumulatoredel riflusso
alimentazione
liquidoW
V'
L'caldaia
D
vapore
LD
fig. 3. Schema di funzionamento di una colonna di distillazione continua.

piatto m. Il bilancio materiale globale relativo alla parte dicolonna sovrastante è espresso da:
[26]
Lo stesso bilancio per il componente i assume la forma:
[27]
dove xD,i è la frazione molare del componente i nella correntedi testa. Combinando le due ultime equazioni si ricava:
[28]
da cui:
[29]
Analogamente si procede per la sezione di esaurimento,isolando una porzione di colonna come schematizzato ancorain fig. 3, dove W indica la portata molare del prodotto di coda.
Se si indica con n un generico piatto della sezione di esau-rimento, il bilancio materiale globale e quello relativo al com-ponente i assumono la forma:
[30]
[31]
Eliminando W si ottiene:
[32]
Le sole equazioni [29] e [32] non sono però sufficienti acaratterizzare il comportamento globale della colonna, e devo-no essere associate a due ulteriori serie di equazioni. Le primestabiliscono un legame tra le composizioni del vapore e delliquido in corrispondenza di un determinato stadio, le secon-de esprimono una relazione fra la portata del liquido che scen-de da un determinato stadio e quella del vapore che sale daquello sottostante.
Ipotizzando che ogni stadio si comporti idealmente e uti-lizzando il rapporto di vaporizzazione, si ottiene:
[33]
La seconda serie di equazioni si formula invece eseguen-do un bilancio entalpico in corrispondenza di ciascun piattodella colonna stessa. Se si considera un piatto generico m e siapplica la [12], tenendo presente che Q e W sono entrambi nulli,si ottiene:
[34]
L’equazione precedente fornisce quindi una relazione trale portate molari dei flussi di vapore e di liquido in corrispon-denza del piatto m.
L’insieme delle relazioni menzionate può essere semplifi-cato. Infatti poiché H��h, ne consegue che nella [34] è legit-timo trascurare il flusso di calore associato alla corrente liqui-da rispetto a quello della corrente vapore, per cui:
[35]
In questa approssimazione il flusso di calore associato conquello del vapore che sale lungo la colonna risulta praticamentecostante. Un’ulteriore approssimazione consiste nel porre:
[36]
Essa risulta accettabile se i calori molari di evaporazionedelle diverse sostanze sono confrontabili tra di loro. Infatti, inquesto caso l’entalpia di una miscela di vapori risulta pratica-mente indipendente dalla sua composizione. Le ultime dueequazioni risultano compatibili solo se:
[37]
Da un bilancio globale sui diversi piatti, se V è costante,risulta inoltre:
[38]
Si assume cioè che sia la portata del liquido sia quella delvapore siano costanti nelle due sezioni della colonna, un’ipo-tesi che viene anche detta ‘del flusso molare costante’.
Le approssimazioni precedenti permettono di semplifica-re in modo significativo l’analisi del comportamento di unacolonna di distillazione, che può essere condotta prescinden-do dal bilancio entalpico su ciascun piatto. Anche le equazio-ni di bilancio materiale risultano così profondamente sempli-ficate. La [29] si può infatti scrivere come segue:
[39]
Se si risolve la precedente rispetto a ym�1, ricordando cheV�L�D, si ottiene:
[40]
Indicando con R il rapporto di riflusso (L /D), la [40] di-venta:
[41]
nota come ‘equazione di lavoro’ della sezione di rettifica.In maniera analoga si sviluppa l’analisi per la sezione di esau-rimento; indicando con L� e V� le corrispondenti portate mola-ri del liquido e del vapore, diverse da quelle nella sezione direttifica, la [39] diventa:
[42]
che, risolta rispetto a yn,i, ricordando che W�V��L, dà:
[43]
detta anche equazione di lavoro della sezione di esaurimento.I valori dei flussi che intervengono nell’ultima equazione pos-sono essere determinati dal bilancio globale della colonna:
[44]
Inoltre, se l’alimentazione è costituita da una corrente liqui-da che si trova alla temperatura corrispondente a quella dellostadio su cui viene alimentata, si può scrivere:
[45]
Nell’ambito delle precedenti approssimazioni è possibile,fruendo delle due equazioni [41] e [43], insieme alle relazio-ni di equilibrio, calcolare il numero di stadi necessari affinchéuna colonna di distillazione possa dare una determinata pre-stazione. Una procedura di calcolo è la seguente: • si fissa la composizione in caldaia xw,i e si determina la
temperatura della caldaia imponendo la condizione:
[46] K T xi w ii
( ) =∑ , 1
L L F RD F�= + = +
F D W= +
y LL W x W
L W xn i n i w i, , ,= − − −+�
� �1
LV
x yx x
w i n i
w i n i
�
�=
−− +
, ,
, ,1
y RR x
xRm i m i
D i+ = + + +1 1 1, ,
,
y L V x D V xm i m i D i+ =( ) +( )1, , ,
LV
x yx xD i m i
D i m i=
−−
+, ,
, ,
1
L L L Lm m m+ − =1 1≈ ≈
V V V Vm m m+ −1 1≈ ≈ =
� � �H H Hm m m+ −1 1≈ ≈ ≈...
� � �H V H V H Vm m m m m m+ + − −1 1 1 1≈ ≈ ≈...
� � � �h L H V V H h Lm m m m m m m m− − + ++ = +1 1 1 1
yx
K T Pi
ii= = ( )Φ ,
LV
x yx x
n
n
w i n i
w i n i
+
+=
−−
1
1
, ,
, ,
V y Wx L xn n i w i n n i, , ,+ = + +1 1
V W Ln n+ = +1
LV
x yx x
m
m
D i m i
D i m i+
+=−−1
1, ,
, ,
V y L x V x L xm i m i m m i m D i m D i+ + += + −1 1 1, , , , ,
V y L x Dxm m i m m i D i+ + = +1 1, , ,
V L Dm m+ = +1
ASPETTI PROCESSISTICI
324 ENCICLOPEDIA DEGLI IDROCARBURI
• si calcola la yw,i tramite la [33];• si valutano le composizioni del liquido che cade dal piat-
to soprastante la caldaia, xi, mediante la [43];• si determina la temperatura della miscela presente sul piat-
to 1 risolvendo l’equazione
[47]
• si calcola quindi la composizione del vapore che sale datale piatto yi e che è in equilibrio con quella del liquido pre-sente sul piatto stesso, facendo nuovamente ricorso alla [33].I passi ora descritti vengono poi applicati allo stadio suc-
cessivo, e si procede iterativamente finché non si incontra unacomposizione del liquido simile a quella della miscela di ali-mentazione.
Un metodo di calcolo analogo può essere applicato allasezione di rettifica, impiegando però l’equazione [40] per cal-colare la composizione del liquido che scende da uno stadiotramite quella del vapore che sale da quello sottostante. Il cal-colo procede finché la composizione del vapore che sale da undeterminato stadio è confrontabile con quella del prodotto ditesta della distillazione. Infatti se la condensazione è totale, lacomposizione del vapore che sale dallo stadio più in alto allasommità della colonna è uguale a quella del prodotto distilla-to, cioè:
[48]
Un bilancio entalpico globale permette infine di calcolarela quantità di calore necessaria per effettuare la distillazione.Risulta:
[49]
dove QC e QD indicano rispettivamente le quantità di calorefornite alla caldaia e sottratte al condensatore per unità di tempo.
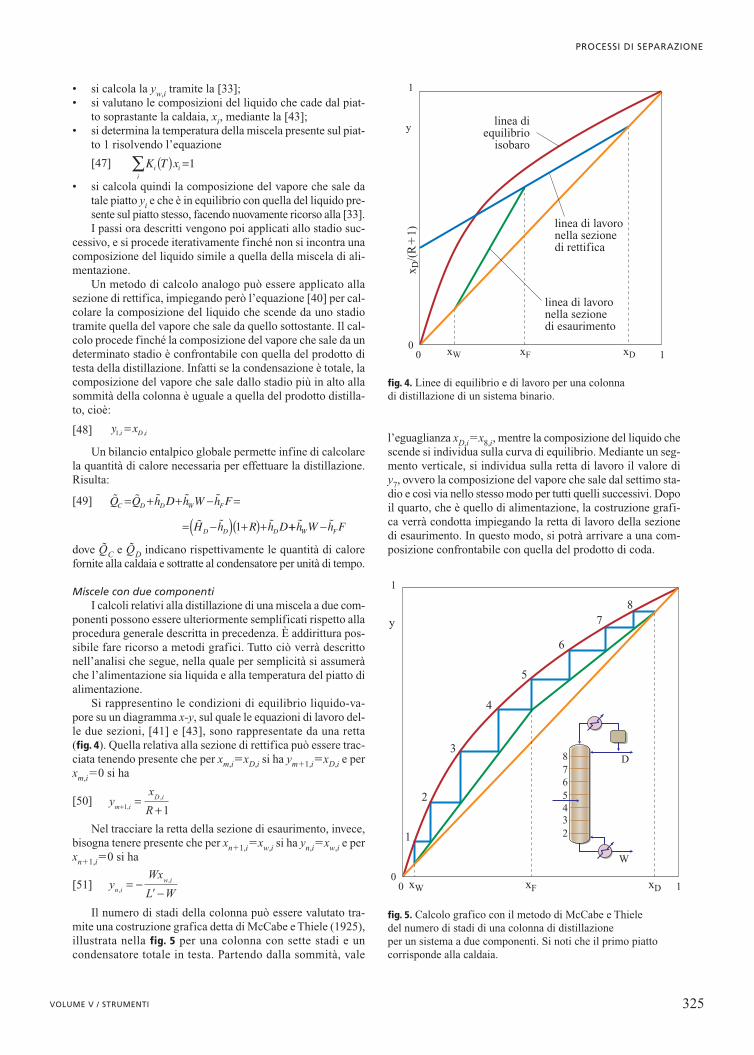
Miscele con due componentiI calcoli relativi alla distillazione di una miscela a due com-
ponenti possono essere ulteriormente semplificati rispetto allaprocedura generale descritta in precedenza. È addirittura pos-sibile fare ricorso a metodi grafici. Tutto ciò verrà descrittonell’analisi che segue, nella quale per semplicità si assumeràche l’alimentazione sia liquida e alla temperatura del piatto dialimentazione.
Si rappresentino le condizioni di equilibrio liquido-va-pore su un diagramma x-y, sul quale le equazioni di lavoro del-le due sezioni, [41] e [43], sono rappresentate da una retta(fig. 4). Quella relativa alla sezione di rettifica può essere trac-ciata tenendo presente che per xm,i�xD,i si ha ym�1,i�xD,i e perxm,i�0 si ha
[50]
Nel tracciare la retta della sezione di esaurimento, invece,bisogna tenere presente che per xn�1,i�xw,i si ha yn,i�xw,i e perxn�1,i�0 si ha
[51]
Il numero di stadi della colonna può essere valutato tra-mite una costruzione grafica detta di McCabe e Thiele (1925),illustrata nella fig. 5 per una colonna con sette stadi e uncondensatore totale in testa. Partendo dalla sommità, vale
l’eguaglianza xD,i�x8,i, mentre la composizione del liquido chescende si individua sulla curva di equilibrio. Mediante un seg-mento verticale, si individua sulla retta di lavoro il valore diy7, ovvero la composizione del vapore che sale dal settimo sta-dio e così via nello stesso modo per tutti quelli successivi. Dopoil quarto, che è quello di alimentazione, la costruzione grafi-ca verrà condotta impiegando la retta di lavoro della sezionedi esaurimento. In questo modo, si potrà arrivare a una com-posizione confrontabile con quella del prodotto di coda.
yWxL Wn i
w i,
,= −−�
yxRm i
D i+ =
+1 1,
,
� � �H h R h DD D D= −( ) +( )+1 ++ −� �h W h FW F
� � � � �Q Q h D h W h FC D D W F= + + − =
y xi D i1, ,=
K T xi ii
( ) =∑ 1
PROCESSI DI SEPARAZIONE
325VOLUME V / STRUMENTI
linea di lavoronella sezionedi rettifica
linea di lavoronella sezionedi esaurimento
linea diequilibrio
isobaro
x D/(
R�
1)
0
1
y
xW xF xD0 1
fig. 4. Linee di equilibrio e di lavoro per una colonna di distillazione di un sistema binario.
y
0
1
1
2
2
3
3
4
4
5
5
6
6
7
7
8
8 D
W
xW xF xD0 1
fig. 5. Calcolo grafico con il metodo di McCabe e Thiele del numero di stadi di una colonna di distillazione per un sistema a due componenti. Si noti che il primo piattocorrisponde alla caldaia.

Se tutto il vapore che sale dall’ultimo stadio dopo la com-pleta condensazione viene rimandato in colonna, si dice che ladistillazione avviene in condizioni di riflusso totale, o infini-to, in cui tutto il vapore condensato viene rinviato nella colon-na stessa. Il rapporto di riflusso R�L �D è infinito, perché D�0,ed entrambe le linee di esercizio si identificano con la diago-nale del diagramma. In questo caso, assegnato il numero dipiatti della colonna, si realizza la massima differenza di con-centrazione del composto in esame tra testa e coda.
Se il punto di intersezione tra le rette di lavoro si trova sullalinea di equilibrio, il numero di piatti necessari per la distilla-zione tende all’infinito, poiché in corrispondenza del punto diintersezione la composizione su ciascun piatto resta pratica-mente costante; il valore di riflusso corrispondente viene chia-mato rapporto minimo di riflusso e si indica con Rm.
Metodi analitici per il calcolo del numero di stadiLa determinazione del numero di stadi di una colonna di
distillazione a due componenti può talvolta essere condottadirettamente per via analitica. Questi metodi in generale por-tano a risultati meno accurati di quelli ottenuti per via graficaprecedentemente illustrati, perché la loro applicazione dipen-de dalla possibilità di descrivere l’equilibrio liquido-vaporemediante una funzione analitica relativamente semplice. Perun sistema binario lo scopo può essere raggiunto impiegandola volatilità relativa:
[52]
che costituisce un caso particolare del rapporto di separazio-ne, definito in [1], corrispondente a un sistema liquido-vapo-re all’equilibrio. Spesso, in particolari intervalli di temperatu-ra alla volatilità relativa a12 si può attribuire un valore a media-mente costante. Per miscele ideali, la cui costante di equilibriosi esprime come:
[53]
dove pi0 rappresenta la tensione di vapore del liquido, la [52]
diventa:
[54]
ed è quindi espressa dal rapporto tra le tensioni di vapore deidue componenti. Ricordando che x1�x2�1 e y1�y2�1, si rica-va la seguente equazione che fornisce la relazione tra y e x,cioè l’andamento delle curve y�f(x) all’equilibrio:
[55]
Applicando questa equazione si possono ricavare le seguen-ti formule che esprimono il numero minimo di stadi della colon-na, escludendo la caldaia, Nm (cioè a riflusso infinito) e Rm(cioè corrispondente a un numero infinito di stadi). La prima,detta formula di Fenske (1932), ha la forma:
[56]
La seconda, invece, detta formula di Underwood (1948),si scrive:
[57]
Le frazioni molari riportate si riferiscono al componentepiù volatile.
Noti i valori di Nm e Rm, si può calcolare approssimativa-mente il numero di stadi della colonna che corrisponde a unvalore generico del rapporto di riflusso tramite la correlazio-ne, detta di Gilliland, costruita su basi empiriche mediante l’a-nalisi di un certo numero di colonne di distillazione in fun-zione. I risultati ottenuti con questo metodo, detto short cuts,sono ragionevolmente accurati e costituiscono una buona basedi partenza per calcoli più dettagliati.
Calcolo rigoroso di una colonna di distillazione a più componenti
Nel seguito sono illustrati i metodi di calcolo applicati perrisolvere i problemi connessi alla simulazione del funziona-mento di una colonna di distillazione continua, con un nume-ro N di componenti e M stadi. Questa situazione si presentafrequentemente nella simulazione degli impianti dell’industriapetrolchimica e petrolifera nella quale si devono trattare misce-le con un numero elevato di componenti, mediante l’impiegodi unità con decine, se non centinaia, di stadi. Tipiche, a que-sto proposito, sono le cosiddette colonne di superfrazionamentoche devono separare composti con temperature di ebollizionemolto simili tra loro, come si verifica per esempio nel casodegli xileni. Dal punto di vista matematico, le equazioni neces-sarie per affrontare questi problemi sono quelle che esprimo-no i bilanci materiali in ciascuno stadio ideale, unitamente allerelazioni termodinamiche che esprimono le condizioni di equi-librio liquido-vapore. Il problema viene solitamente affronta-to simulando il comportamento di una colonna di cui sianoassegnati il numero di stadi e le condizioni di esercizio. I risul-tati del calcolo devono fornire le temperature e la composi-zione in ciascun stadio, oltre che le portate e le composizionidelle correnti uscenti dalla colonna stessa. Pertanto, dal puntodi vista matematico il problema si presenta ben definito mapiuttosto complesso, soprattutto quando i valori di N e M sonoelevati. In realtà la disponibilità di grandi calcolatori ha resopossibile l’esecuzione di calcoli rigorosi i cui limiti, allo statoattuale, si devono essenzialmente attribuire all’accuratezzadelle informazioni disponibili sulle proprietà chimico-fisichedelle sostanze e delle miscele in gioco.
Per conferire maggiore generalità al problema, è opportu-no tenere presente che spesso si effettua l’immissione o il pre-lievo di miscele su alcuni piatti della colonna, poiché questoprocedimento agevola la separazione di particolari tagli dellamiscela aventi la composizione desiderata. Pertanto per cia-scuno stadio viene contemplata la possibilità che possa esserepresente una alimentazione, o un prelievo, o uno scambio dicalore. Oltre ai simboli già introdotti, nel seguito si indicanocon Fn l’alimentazione sullo stadio n, con zn,i la composizionedell’i-esimo componente in F, con wn il vapore prelevato dallostadio n, con Un il liquido prelevato dallo stadio n e con Qn ilcalore scambiato sullo stadio n. Ovviamente i valori di questegrandezze saranno diversi da zero solo per pochi stadi, poichésolo su alcuni di essi può avere luogo lo scambio con l’ester-no di materia e di calore.
La colonna presa in esame ha la configurazione indicatanella fig. 6 ed è costituita da N stadi includenti un condensato-re, parziale o totale (stadio 1), e una caldaia (stadio N). Tutti ivalori delle grandezze precedentemente menzionate devono esse-re noti, unitamente alle condizioni di equilibrio liquido-vapore.
Ciò premesso, è possibile dimostrare che se si effettua ilbilancio materiale di ciascun componente su ciascuno stadio,
Rxx
xxm
D
F
D
F
=−
−−−
1
1
1
1αα
N
x xx x
m
D m
w D+ =
−( )−( )1
11
ln
lnα
y xx1
12 1
1 12 1 1=
−( )+αα
α12 10
20= p p
K pPii=0
α121 2
2 1= y xy x
ASPETTI PROCESSISTICI
326 ENCICLOPEDIA DEGLI IDROCARBURI

si ricava una serie di equazioni che sinteticamente si possonoesprimere come segue:• stadio 1:
[58]
• stadi 2�n�N�1:
[59]
• stadio N:
[60]
dove le espressioni specifiche dei parametri presenti nelle equa-zioni sono riportate nella tab. 2. Inoltre, se si pone Wn�Ln+Un,si dimostra che i bilanci energetici condotti su ciascuno stadioassumono la forma:
[61]
Le precedenti costituiscono un sistema di equazioni alge-briche, (N�1)M esprimenti i bilanci materiali su ciascuno sta-dio e M esprimenti i bilanci energetici. A esse si debbono aggiun-gere le seguenti 2N equazioni:
[62]
[63]
dove la [62] consente di valutare la temperatura di ebollizionedel liquido e la [63] la temperatura di condensazione del vapore.
Pertanto, globalmente si tratta di NM�2N equazionicon un corrispondente numero di incognite, precisamente NM
esprimenti le composizioni del liquido presente su ciascunostadio e 2N esprimenti le temperatura del liquido e del vaporesu ciascuno stadio.
La dipendenza delle K (T, p, xi, yi) dalle diverse variabilicoinvolte nel processo conferisce un carattere non lineare alleequazioni precedenti. Pertanto la soluzione del sistema puòessere ottenuta solo per via numerica, fruendo di procedimen-ti di calcolo iterativi. Esistono diversi approcci per affrontarequesto problema, che si riconducono ai metodi numerici disoluzione dei sistemi di equazioni algebriche non lineari conle caratteristiche tridiagonali come quello in esame. Si posso-no, per esempio, impiegare processi iterativi mediante i qualisi modificano, in passi di calcolo successivi, valori ipotizzatidelle temperature, delle composizioni e dei flussi del liquidoe del vapore. Alternativamente si può impiegare la tecnica diNewton-Raphson per linearizzare le equazioni in ogni stadiodel calcolo. In entrambi i casi si fruisce di formule ricorrentiche possono essere impiegate per la soluzione di sistemi diequazioni lineari.
Metodi globali approssimati per sistemi a più componentiAnche per sistemi a più componenti esistono metodi
approssimati per la determinazione del numero di stadi, basa-ti sempre sull’assunzione che le volatilità relative dei varicomponenti possano essere ritenute costanti. Solitamente que-sti metodi si basano sull’individuazione di due componentichiave nell’alimentazione. Il componente chiave leggero è ilpiù volatile dei componenti di coda, mentre il componentechiave pesante è il meno volatile tra quelli di testa. L’equa-zione di Fenske [56] può quindi essere applicata facendo rife-rimento ai componenti chiave, ricavando così il numero mini-mo di stadi Nm:
[64]
dove xL e xP indicano rispettivamente le frazioni molari dei com-ponenti chiave leggero e pesante, mentre gli indici D e W indi-cano i prodotti di testa e di coda.
Un calcolo approssimato del rapporto di riflusso può esse-re eseguito applicando le seguenti equazioni di Underwood:
N
xx
xx
m
L
P D
P
D W
LF+ =
1ln
lnα
yK xi
n iii
i,∑ ∑= =1
K x yn i n i iii
, , = =∑∑ 1
� �h hn n−+ − 1(( ) + −( )+−L F h H Qn n n F n n1� �
,
� � � �H h V H h V Wn n n n n n n+ +−( ) = −( ) +( )+1 1
A x B xN N i N N i− + =1 0, ,
A x B x C x F zn n i n n i n n i n n i− ++ + + =1 1 0, , , ,
B x C xi i1 1 1 1 0, ,+ =
PROCESSI DI SEPARAZIONE
327VOLUME V / STRUMENTI
V2
V1
U1
f2
fn
fW
fD
fn�1
V2
V3
Vn
VH
U2
Un�1
Ln�1
Un
L1
Ln
W2
W3
Wn
W�LN�UN
D�
V1�
U1
Wn�1
Vn�1
F2
F3
Fn
Fn�1
fig. 6. Schema di una colonna di rettifica a più componenti.
tab. 2. Espressioni dei parametri delle equazioni [58-60]
B V K U Li1 1 1 1 1= − + +( ),
A L V F W U Dn n n k k kk
n
= = + − −( ) −−=
−
∑12
1
B V W K V F W U D Un n n n i n i k k k nk
n
= − +( ) + + − −( ) − + =
∑, ,2
D V U= +1 1
A V WN N= +
B V K WN N N i= − +( ),
W L UN N= +
WN = 0
C V Kn n n i= + +1 1,
(Kn�1 è la costante di equilibrio sul piatto i)

[65]
dove ai rappresenta la volatilità relativa del componente i rispet-to a uno generico assunto come riferimento. Sviluppando laprima relazione si ottiene un’equazione di grado n (pari alnumero di componenti) in funzione della variabile ÿ. Tra lesoluzioni dell’equazione bisogna scegliere quella compresa trale volatilità relative dei componenti chiave. Sostituendo il valo-re così ottenuto nella seconda delle [65], si ricava il valore diRm. Dai valori di Nm e Rm ottenuti in questo modo si risale poial numero di piatti corrispondenti a un dato riflusso mediantela correlazione di Gilliland.
Trasferimento di materia negli stadi di un’apparecchiatura vapore (gas)-liquido
La trattazione fin qui esposta dei processi di distillazioneè basata sull’impiego di opportuni stadi nei quali sono pre-senti le due fasi liquido e vapore in intimo contatto, in modotale da agevolare i mutui processi di trasporto di materia. Inparticolare, gli stadi possono essere realizzati con i menzio-nati piatti forati, anche se si è rivelato estremamente efficien-te l’impiego di particolari riempimenti detti ‘strutturati’ (v.oltre). Prescindendo per ora da questo aspetto, risulta utile svi-luppare l’analisi delle colonne di distillazione, assumendo inprima istanza che ciascuno stadio si comporti idealmente, poi-ché le correnti di vapore e di liquido che si separano da essosi trovano in condizioni di equilibrio termodinamico. In pra-tica ovviamente ciò non si verifica, poiché la composizionedel vapore che lascia lo stadio è meno ricca dei componentivolatili di quanto previsto dalle condizioni di equilibrio, soprat-tutto poiché il tempo di contatto tra le due fasi non è in gene-rale sufficiente per raggiungere l’equilibrio. In sostanza, perpoter valutare la corretta composizione delle due fasi in gioco,è necessario estendere l’analisi sino a ora condotta, tenendoesplicitamente conto dell’influenza della velocità con la qualehanno luogo i processi di trasferimento di materia fra le fasiliquido e vapore presenti nello stadio stesso. Ovviamente essadipenderà sia dalle caratteristiche geometriche del piatto o delriempimento, sia da quelle fluidodinamiche che si instauranosu di esso.
Per esprimere la velocità con cui hanno luogo i processi ditrasferimento di materia tra due fasi a contatto (vapore-liqui-do nel caso della distillazione e gas-liquido nel caso dell’as-sorbimento; v. par. 6.2.4), si può fare ricorso alla teoria del dop-pio film, in base alla quale si assume che nell’ambito di ognifase la velocità di trasferimento di ciascun componente sia pro-porzionale alla differenza fra la sua pressione parziale o con-centrazione nel cuore della fase in esame e il suo valore allasuperficie interfasica (identificata con il pedice i), ovvero:
[66]
[67]
dove N1 rappresenta le moli della specie 1 che vengono tra-sferite per unità di tempo e per unità di superficie di contatto,kg e kc sono opportuni coefficienti, detti di trasporto di mate-ria, i cui valori dipendono prevalentemente dalle menzionatecaratteristiche fluidodinamiche che si instaurano su un piatto,mentre le concentrazioni C1 e C1i sono generalmente espressein moli della specie 1 per unità di volume.
Se si suppone che alla superficie interfasica sussistano con-dizioni di equilibrio, si può porre:
[68]
essendo Hi un opportuno parametro mediante il quale risultapossibile esprimere l’equilibrio fra le due fasi attraverso unarelazione lineare.
In condizioni stazionarie le velocità di trasporto di mate-ria in entrambe le fasi devono essere uguali
[69]
da cui:
[70]
Noti quindi kc e kg, è possibile ricavare p1i e C1i risolven-do le due equazioni [68] e [70].
Tuttavia è in generale più conveniente esprimere la velo-cità del processo di trasferimento di massa, utilizzando gran-dezze operativamente misurabili come concentrazioni o pres-sioni parziali presenti nel cuore delle due fasi. Risulta quindiopportuno introdurre due nuovi coefficienti globali di trasfe-rimento di materia definiti come:
[71]
dove p1* rappresenta la pressione parziale che si avrebbe nel gas
se fosse in equilibrio con C1, mentre C1* è la concentrazione
che si avrebbe nel liquido se fosse in equilibrio con p1. Ovvia-mente valgono le relazioni:
[72]
Si ricava allora che:
[73]
e
[74]
L’applicazione delle equazioni precedenti presume, comeè stato anticipato, che siano note le caratteristiche geometrichee fluidodinamiche dei piatti impiegati nel processo, che ovvia-mente risultano necessarie per poter valutare i coefficienti ditrasporto di materia delle due fasi individuali. Il problema ècomunque di notevole complessità e viene affrontato fruendodi opportuni modelli di flusso. Per esempio, in un’impostazio-ne semplificata si assume che entrambe le fasi, liquido e vapo-re, siano perfettamente rimescolate, per cui le concentrazionidei diversi componenti risultano uniformi sull’intero piatto.
Prescindendo da analisi più dettagliate che permettono didescrivere con ragionevole accuratezza le deviazioni dall’i-dealità che si instaura sui piatti di una colonna di distillazio-ne, è importante ricordare che lo scostamento rispetto alle con-dizioni di equilibrio viene solitamente espresso tramite un para-metro globale, chiamato efficienza del piatto, o di Murphree(1925), definita dalla seguente relazione:
[75] Ey yy yMVn n
n n
=−−
+∗
+
1
1
1 1 1
K m k kc i g c
= +
1 1K k
mkg g
i
c
= +
p f C
p f C1 1
1 1
= ( )= ( )
∗
∗
N K p p
N K C Cg
c
1 1 1
1 1 1
= −( )= −( )
∗
∗
p pC C
kk
i
i
c
g
1 1
1 1
−− =−
k p p k C C k C Cg i c i c i1 1 1 1 1 1−( )= −( )=− −( )
p f C H Ci i i i1 1 1= ( )=
N k C Cc i1 1 1= −( )(per la fase liquido)
N k p pg i1 1 1= −( )(per la fase gas o vapore)
αα ϑαα ϑ
i F i
ii
i D i
iim
x
xR
,
,
− =
− = +
∑
∑
0
1
ASPETTI PROCESSISTICI
328 ENCICLOPEDIA DEGLI IDROCARBURI

dove yn* indica la composizione in equilibrio con la composi-
zione xn del liquido che lascia il piatto e yn è la composizionedel vapore che lascia il piatto.
6.2.4 Assorbimento di gas in liquidi
L’assorbimento è l’operazione nel corso della quale una misce-la gassosa viene portata a contatto con un liquido, con l’intentodi separarne uno dei suoi componenti sciogliendolo nel liqui-do stesso. Esistono diversi esempi industriali nei quali si attuaquesta operazione; di particolare interesse è la rimozione delbiossido di carbonio dal gas di sintesi e dai prodotti di com-bustione, attraverso il lavaggio con acqua sotto pressione, consoluzioni di etanolammine o con altri agenti specifici. Un ulte-riore esempio riguarda il lavaggio del gas di un forno a cokeche viene prima trattato con acqua per rimuovere l’ammonia-ca, e poi con un olio minerale per togliere i vapori di benzenee toluene. L’obiettivo dell’assorbimento può essere quindi lapurificazione dei gas, ma anche il recupero di prodotti o la pro-duzione di adeguate miscele di gas.
La scelta del liquido più opportuno per effettuare un’ope-razione di assorbimento è generalmente guidata dai criteri diseguito elencati.
Solubilità dei gas. Dovrebbe essere elevata, in modo daaumentare la velocità del processo e diminuire le quantità disolvente richieste. Si può ottenere una buona solubilità usan-do solventi la cui natura chimica sia simile a quella del solutoche deve essere rimosso. Se tra solvente e soluto avviene unareazione chimica si ottiene una solubilità molto elevata, maaffinché il solvente possa essere recuperato la reazione deveessere reversibile.
Volatilità. Il solvente dovrebbe avere una tensione di vapo-re bassa, dal momento che il gas uscente da un assorbimentoè normalmente saturo di solvente, per cui si rischia di perder-ne quantità consistenti. Talvolta si usa anche un secondo liqui-do meno volatile per recuperare la porzione evaporata del primo.
Bassa corrosività. I materiali richiesti per la costruzionedell’impianto non devono necessariamente essere molto resi-stenti alla corrosione, al fine di abbassare il costo totale.
Costo del solvente. Deve essere basso, in modo che even-tuali perdite non risultino onerose.
Viscosità del liquido solvente. Deve essere bassa per aumen-tare le velocità di assorbimento, minimizzare le perdite di cari-co e migliorare le proprietà di scambio termico.
Altre caratteristiche. Il solvente dovrebbe essere atossico,chimicamente stabile e avere una bassa temperatura di conge-lamento.
Solitamente l’operazione di assorbimento viene condottain colonne verticali in cui sono presenti due flussi in contro-corrente, uno del gas dal basso verso l’alto e l’altro del liqui-do nella direzione opposta. Il contatto tra le due fasi viene rea-lizzato mediante stadi o più comunemente in modo continuolungo tutta la colonna, utilizzando corpi di riempimento comeillustrato in fig. 7. Le condizioni di lavoro, o esercizio, di unacolonna di assorbimento sono derivate dal suo bilancio mate-riale. Si può supporre che la miscela di alimentazione sia costi-tuita da un componente solubile e uno insolubile in un liquidosolvente, di cui si può trascurare l’evaporazione. In questo casoi flussi molari del gas insolubile e del solvente vengono rite-nuti costanti lungo tutta la colonna.
Indicando con Y la concentrazione nel gas del componen-te solubile, espressa in moli di gas/mole di gas insolubile, e
con X la concentrazione nel liquido del componente solubileespressa in moli/mole di solvente, ovviamente si ha X�x�(1�x)e Y�y�(1�y), dove x e y sono rispettivamente le frazioni mola-ri del componente solubile nel gas e nel liquido. Indicando poicon Gs la portata molare del gas insolubile per sezione unita-ria di colonna e con Ls la portata molare del solvente puro perla sezione unitaria di colonna, il bilancio materiale dell’interacolonna riferito al componente solubile ha la forma seguente:
[76]
mentre tra un estremo della colonna, per esempio la base, euna sezione generica il bilancio materiale è dato da:
[77]
ovvero da:
[78]
Quest’ultima relazione esprime il legame che intercorre trala frazione molare del componente solubile nella miscela gas-sosa e la corrispondente frazione molare nella fase liquida.Riportando su un diagramma l’andamento di y in funzione dix, si ottiene una curva, detta linea di lavoro o di esercizio, chedeve essere situata al di sopra della linea di equilibrio, poichésolo a tale condizione la concentrazione del componente so-lubile nella fase gassosa è superiore a quella che corrispon-derebbe alle condizioni di equilibrio, determinando quindi un
G yy
yy L x
xxxs s
1
1
1
11 1 1 1− − −
= − − −
G Y Y L X Xs s1 1−( )= −( )
G Y Y L X Xs s1 2 1 2−( )= −( )
PROCESSI DI SEPARAZIONE
329VOLUME V / STRUMENTI
z
dzZ
G2,y2
G1,y1
L2,x2
L1,x1
liquido
gas
fig. 7. Schema di una colonna di assorbimento gas-liquido. Nel particolare è mostrata una parte del riempimento formata da anelli Raschig, bagnati dal liquido e percorsi in controcorrente dal gas.

passaggio di tale componente dalla fase gassosa a quella liquida.Si ricorda che la linea di equilibrio esprime, nelle condizioni ditemperatura e pressione a cui opera la colonna, la relazione cheintercorre tra y e x in condizioni di equilibrio termodinamico;quando si ha a che fare con gas diluiti la relazione di equilibriopuò essere espressa fruendo della legge di Henry, in base allaquale, a temperatura costante, la pressione parziale di un com-ponente nella fase gassosa viene assunta proporzionale alla suaconcentrazione molare attraverso la costante di equilibrio H.
Se l’operazione di assorbimento viene condotta in una colon-na a più stadi, il loro numero viene valutato attraverso una costru-zione grafica simile a quella di McCabe e Thiele descritta per ilcalcolo degli stadi di una colonna di distillazione. In una colon-na a riempimento la velocità con cui ha luogo il processo di assor-bimento si calcola per esempio mediante la prima delle equa-zioni [71], in cui la forza motrice del processo è data dalla dif-ferenza tra la pressione parziale del componente solubileeffettivamente presente nella fase gassosa e la pressione parzia-le che esso avrebbe se fosse in equilibrio con la fase liquida.
Si consideri ora un elemento di colonna di altezza dz e areaA (v. ancora fig. 7). Indicando con a la superficie di contattoliquido/gas per volume unitario di colonna, le moli di gas tra-sferite nell’unità di tempo dalla fase gassosa alla fase liquidanell’elemento di volume considerato sono date, ricordando la[71], dalla relazione:
[79]
Eguagliando il secondo membro di questa espressione conquella del bilancio materiale effettuato sull’elemento di volu-me in questione, si ottiene:
[80]
Isolando dz e integrando l’espressione così ottenuta tra ledue estremità della colonna, si ricava l’altezza totale Z dellacolonna necessaria per realizzare la separazione:
[81]
Se la concentrazione del componente solubile nella fasegassosa è piccola, è legittimo assumere che (1�y)2�1 e Gs�G,per cui l’equazione precedente diventa:
[82]
Solitamente l’integrale al secondo membro della prece-dente equazione viene chiamato numero di unità di trasferi-mento, e viene indicato con NOG. Si noti infatti che se y�y* èmediamente piccolo, NOG risulta elevato, mentre, al contrario,se tale differenza è elevata NOG è un numero piccolo. In altritermini, NOG esprime la difficoltà che si incontra nel realizza-re l’operazione. La [81] si può quindi scrivere in questo modo:
[83]
dove l’espressione HOG�G�KgaP, che ha le dimensioni di unalunghezza, viene detta altezza di una unità di trasferimento,HTU (Height of a Transfer Unit). Se si riscrive l’equazione[73] ponendo Hi uguale alla costante di Henry e moltiplican-do i vari termini per G�aP, si ottiene:
[84]
In quest’ultima espressione si è tenuto conto del fatto chela costante di Henry si può scrivere nella forma H�mP/C, dovem rappresenta la costante di equilibrio nel caso in cui si adot-tino le frazioni molari per esprimere le concentrazioni del com-ponente solubile; C è espresso in moli totali per unità di volu-me. Si può allora decomporre l’unità di trasferimento in duecontributi, ciascuno relativo alle fasi presenti nel sistema, tra-mite la relazione:
[85]
essendo HG�G�kgaP e HL�L�kcaC.
Apparecchiature di contatto gas-liquidoIn entrambe le operazioni di distillazione e assorbimento
si utilizzano particolari dispositivi, quali i piatti o i corpi diriempimento, per realizzare il contatto necessario al trasferi-mento di materia, calore e quantità di moto tra le varie fasicoinvolte. Come già menzionato, l’esempio tipico di piatto èquello forato nel quale il liquido si sposta trasversalmente equindi scende nel piatto inferiore attraverso un condotto, dopoaver superato lo stramazzo di un troppo pieno (fig. 8), mentreil vapore che sale in controcorrente viene fatto gorgogliare nelliquido. Il contatto che si realizza su ciascun piatto tra liquidoe vapore permette lo scambio tra i componenti più volatili chesi accumulano nella fase vapore e i meno volatili che si accu-mulano nella fase liquida. Il flusso del vapore attraverso unaserie di fori esistenti sulla superficie del piatto ne garantisceun efficace gorgogliamento. Per impedire che il liquido discen-da dai fori è necessario che la velocità del vapore sia superio-re a un certo valore limite.
La dispersione del vapore nel liquido su un piatto si puòanche realizzare mediante opportuni dispositivi come le cam-
H H mGL
HOG G L= +
GK aP
Gk aP
H Gk aP
Gk aP
mGL
Lk aCg g
i
c g c= + = +
Z GK aP N H N
gOG OG OG= =
Z GPK a
dyy yg y
y=
− ∗∫2
1
Z GPK a
dyy y y
s
g y
y=
−( ) −( )∗∫ 1 21
2
AG yy
AG dy
yPK y y aAdzs s gd
1 12−
=
−( )= −( )∗
NaAdz PK y y aAdzg= −( )∗
ASPETTI PROCESSISTICI
330 ENCICLOPEDIA DEGLI IDROCARBURI
liquido provenientedal piatto superiorestramazzo
vapore provenientedal piatto inferiore
schiuma
fig. 8. Schema di funzionamento di un piatto forato.

panelle (a bolle oppure a valvola). Ovviamente il buon fun-zionamento del piatto richiede che la velocità del vapore, o delgas, sia opportunamente calibrata in modo da limitare il tra-scinamento di gocce del liquido e di garantire nel contempo lasua stabilità.
Negli impianti con corpi a riempimento, invece, il contat-to gas-liquido è realizzato riempiendo la colonna con oppor-tuni materiali, come quelli illustrati in fig. 9. Le loro dimen-sioni medie sono di 12-50 mm. Nella parte superiore dellacolonna viene alimentato il liquido mediante opportuni distri-butori che ne garantiscono una buona dispersione. Alla basedella colonna si alimenta il gas che fluisce in controcorrenteal liquido. Dal punto di vista fluidodinamico le colonne a riem-pimento devono soddisfare particolari vincoli, perché non abbialuogo un loro totale riempimento di liquido (allagamento oflooding). Nel contempo si deve tenere presente che entrambele portate del gas e del liquido influiscono in modo significa-tivo sui valori dei coefficienti di trasferimento di materia tra lefasi. Questo aspetto è stato l’oggetto di estese indagini sia spe-rimentali sia teoriche, grazie alle quali sono state formulateopportune correlazioni che permettono di valutare i valori deglistessi coefficienti di trasferimento di materia.
In questi ultimi anni stanno trovando ampia diffusione isistemi con riempimenti strutturati, spesso impiegati anche peril revamping di colonne già esistenti. Solitamente sono costi-tuiti da fogli corrugati di metallo, oppure di materiale plasticoo ceramico. I fogli adiacenti sono perpendicolari tra loro inmodo da formare una struttura a nido d’ape molto aperta, chedia luogo a canali di flusso inclinati con aree superficiali ele-vate, grazie alle quali si realizza un intimo contatto tra le duefasi coinvolte, attraverso diverse possibili configurazioni. Que-sto tipo di soluzione presenta svariati vantaggi, in particolaresi hanno piccole cadute di pressione e un efficace contatto frale fasi, dovuti al fatto che l’orientamento verticale dei fogli cor-rugati permette di eliminare ogni superficie orizzontale checrei resistenza al flusso del fluido. Inoltre i riempimenti strut-turati permettono di evitare la creazione di canali preferenzia-li per il passaggio di una delle fasi, fenomeno abbastanza comu-ne nei sistemi convenzionali con corpi di riempimento, e diottenere quindi un significativo miglioramento dell’efficienzadi trasferimento di materia. Tuttavia bisogna tenere presente
che i riempimenti strutturati possono presentare problemi confluidi che danno luogo a fenomeni di sporcamento, poiché laloro pulizia è ovviamente difficoltosa. Inoltre il comportamentodei riempimenti strutturati in operazioni ad alta pressione si èrivelato decisamente meno soddisfacente, poiché al cresceredella densità del vapore può accadere che il liquido venga spin-to verso l’alto dal vapore, provocando retromiscelazioni e con-seguenti riduzioni di capacità e di efficienza.
L’impiego di sistemi di riempimento nelle colonne di distil-lazione crea il problema della loro simulazione che deve esse-re formulata sulla base delle equazioni di bilancio di ciascuncomponente condotto su ogni stadio elementare della colonnastessa e integrato quindi su tutta la sua altezza.
Alternativamente, si può procedere attraverso la valuta-zione del numero di stadi teorici, fruendo successivamente delconcetto di altezza equivalente di uno stadio teorico HETP(Height Equivalent to a Theoretical Plate), che costituisce l’al-tezza di riempimento che nelle identiche condizioni operativesvolge lo stesso lavoro di uno stadio ideale. Si tratta di un para-metro che ovviamente dipende dalla natura e dalle caratteristi-che del riempimento, dalle particolarità dei fluidi in gioco edalle condizioni fluidodinamiche nella colonna stessa. Tuttiquesti fattori vengono compendiati in opportune correlazionisemiempiriche che, nel caso dei riempimenti strutturati, pro-vengono dalle ditte fornitrici dei riempimenti. In realtà poichéi parametri sulla cui base viene espressa l’HETP sono gli stes-si impiegati per esprimere i coefficienti di trasferimento di mate-ria, i due metodi descritti si possono considerare equivalenti.
6.2.5 Estrazione liquido-liquido
L’estrazione con solventi, o estrazione liquido-liquido, è un’o-perazione che permette di separare un componente presente inuna miscela liquida attraverso l’impiego di un solvente, grazieal quale i diversi componenti della miscela originale presenta-no diversi gradi di solubilità. Un semplice esempio permettedi specificare il suo scopo e alcune delle sue caratteristiche.Se una soluzione acquosa di acido acetico viene portata a con-tatto con un opportuno liquido, per esempio un estere qua-le l’acetato di etile, e agitata, una certa quantità di acido si
PROCESSI DI SEPARAZIONE
331VOLUME V / STRUMENTI
A B C
D E F
fig. 9. Materiali di riempimento. A, anelli Raschig; B, anelli Lessing; C, sellettedi Berl; D, sellette Intalox; E, tellerette; F, anelli di Pall.

trasferisce nell’estere. Poiché le densità dello strato acquoso edell’estere sono diverse, terminata l’agitazione le due fasi siseparano. La fase acquosa risulta così depauperata del com-ponente solubile che viene separato da essa. L’acqua residuapuò essere trattata ripetutamente con l’estere, in modo da ridur-re, attraverso stadi successivi, la quantità di acido. Tutto ciòpuò essere realizzato in una cascata di stadi in controcorrente,oppure in un’apparecchiatura che lavori per contatto diretto,sempre in controcorrente. Anche in questo caso, come nelladistillazione, l’uso di un riflusso può contribuire a migliorareil grado finale di separazione. A fine operazione il solventepuò essere recuperato tramite distillazione.
Affinché sia possibile applicare questo metodo di separa-zione è necessario operare in un campo di composizioni taliper cui nel sistema siano presenti due fasi liquide, una ricca insolvente chiamata estratto (E) e l’altra chiamata raffinato (R).Talora si agisce su miscele a più componenti, ma per sempli-cità nel seguito si suppone di avere a che fare con sistemi incui si hanno esclusivamente le seguenti fasi: la fase estratta checontiene il componente di interesse, o soluto, nel solvente; lamiscela da raffinare; il solvente selettivo per il componente daestrarre.
Spesso l’estrazione con solventi viene impiegata in sosti-tuzione della distillazione, particolarmente quando le sostan-ze da separare sono piuttosto diverse dal punto di vista chimi-co. In questo caso è importante valutare i costi dell’operazio-ne, tenendo conto del fatto che l’estrazione produce una nuovasoluzione che deve essere a sua volta purificata per distilla-zione. Per esempio risulta difficile separare l’acido acetico insoluzione acquosa diluita, mentre è più semplice utilizzareun’estrazione seguita dalla distillazione dall’estratto, tanto piùconveniente quanto più diluita è la soluzione, laddove occor-rerebbe separare grandi quantità d’acqua. Un altro caso in cuil’estrazione rappresenta un’alternativa importante alla distil-lazione è quello in cui si devono trattare sostanze termicamenteinstabili e che quindi non possono essere sottoposte alle tem-perature relativamente elevate richieste per la distillazione:questo caso è tipico dell’estrazione della penicillina e di moltealtre sostanze dell’industria farmaceutica.
Talvolta, invece, l’estrazione rappresenta l’unica soluzio-ne praticabile, non essendo possibile ottenere la separazionein modo alternativo. Per esempio la separazione di idrocarbu-ri paraffinici e aromatici con pesi molecolari simili è moltodifficile se condotta per distillazione, dato che le loro tensio-ni di vapore hanno valori molto vicini, ma può essere ottenu-ta con relativa facilità impiegando una serie di solventi, qualiil biossido di zolfo o il glicole dietilenico.
Come menzionato, l’estrazione liquido-liquido può esse-re realizzata con un procedimento a stadi oppure tramite uncontatto continuo. Nel primo caso la miscela da raffinare F eil solvente S vengono tenuti in intimo contatto mediante un’a-gitazione efficace in modo da far sì che il sistema si avvicinialle condizioni di equilibrio termodinamico. Nella fig. 10 vieneillustrato lo schema di uno stadio di estrazione. Nel miscela-tore M avviene un’agitazione che provoca la dispersione di unodei due liquidi in forma di piccole gocce (fase dispersa) immer-se nell’altro liquido (fase continua), così da accelerare il pro-cesso di trasferimento di materia tra le due fasi. La miscelapassa poi nel decantatore D dove permane per il tempo neces-sario affinché le due fasi formino i due strati E ed R che ven-gono successivamente separati.
Un processo a contatto continuo nella sua forma più sempli-ce si realizza invece facendo fluire, perlopiù in controcorrente,
i due liquidi in torri verticali. Il flusso dei due liquidi vieneottenuto per gravità sfruttando la differenza tra i loro pesi spe-cifici.
I solventi impiegati nelle operazioni liquido-liquido soli-tamente soddisfano le seguenti caratteristiche: a) capacitàdi sciogliere in quantità elevata la specie che deve essereseparata; b) selettività nei confronti del componente da sepa-rare; c) sufficiente volatilità in modo da poter essere facil-mente distillati nella successiva fase di recupero; d ) bassocosto, atossicità e non corrosività; e) bassa viscosità in mododa poter fluire agevolmente nei condotti dell’impianto diestrazione.
Ovviamente nessun solvente soddisfa contemporaneamentetutte le caratteristiche menzionate, per cui la scelta di un sol-vente opportuno implica un compromesso tra i diversi fattori.
Il fattore di separazione definito dalla [1] per l’estrazioneliquido-liquido ha la seguente forma:
[86]
dove gli indici a e b indicano le due fasi, mentre i g sono icoefficienti di attività dei due componenti nelle due fasi, eSa
ij�gia�gj
a è un parametro chiamato selettività della fase.In molti casi la selettività nei processi di estrazione si deve
attribuire alla capacità del solvente di dare legami idrogenocon le molecole che vengono estratte, o di dare luogo, median-te interazioni chimiche, alla formazione di addotti che rendo-no una determinata sostanza preferenzialmente solubile in unparticolare solvente. Per esempio il dimetilenglicole manife-sta proprietà selettive nell’estrazione degli idrocarburi aroma-tici attraverso la formazione di legami idrogeno. La solubilitàdegli idrocarburi nel riguardo di diversi solventi è stata ogget-to di ampi studi.
Estrazione a stadiUn processo di estrazione liquido-liquido a stadi viene
attuato impiegando una batteria di stadi in serie con una dispo-sizione in controcorrente. Ciascun estrattore viene alimenta-to con due correnti, quella del raffinato e quella dell’estratto.Un problema importante è la determinazione del numero distadi necessari per far sì che la composizione della sostanzache si desidera estrarre nella corrente F scenda al di sotto diun certo valore.
A scopo esemplificativo verrà considerato il caso appros-simato in cui il solvente è praticamente immiscibile nel raffi-nato. In tale caso è opportuno indicare con A la massa deldiluente presente nella corrente di alimentazione (F) e con Bla massa del solvente puro presente nella corrente S. Si sup-pone inoltre che la sostanza da estrarre si ripartisca tra le duefasi attraverso una relazione di equilibrio del tipo K�X/Y, nellaquale si possa attribuire alla costante di ripartizione un valore
αγ γγ γ
α β
β α
α β
β α
α
βiji j
i j
i j
i j
ij
ij
x xx x
SS
= = =
ASPETTI PROCESSISTICI
332 ENCICLOPEDIA DEGLI IDROCARBURI
F
SM
D
E
R
fig. 10. Schema di un’apparecchiatura di estrazione a uno stadio.
mediamente costante, dove X�(massa di sostanza)/(massa disolvente) e Y�(massa di sostanza)/(massa di diluente).
In questo caso i bilanci materiali tra i diversi stadi si pos-sono scrivere come segue:• stadio 1:
[87]
ovvero: Y2�Y1�(BK�A)Y1• stadio 2:
[88]
ovvero: Y3�Y2�(BK�A)(Y2�Y1)�(BK�A)2Y1• stadio N:
[89]
ovvero: Yn�1�Yn�(BK�A)(Yn�Yn�1)�(BK�A)nY1Sommando membro a membro tutte le espressioni prece-
denti si ottiene:
[90]
Il termine tra parentesi è una progressione geometrica, percui si ottiene:
[91]
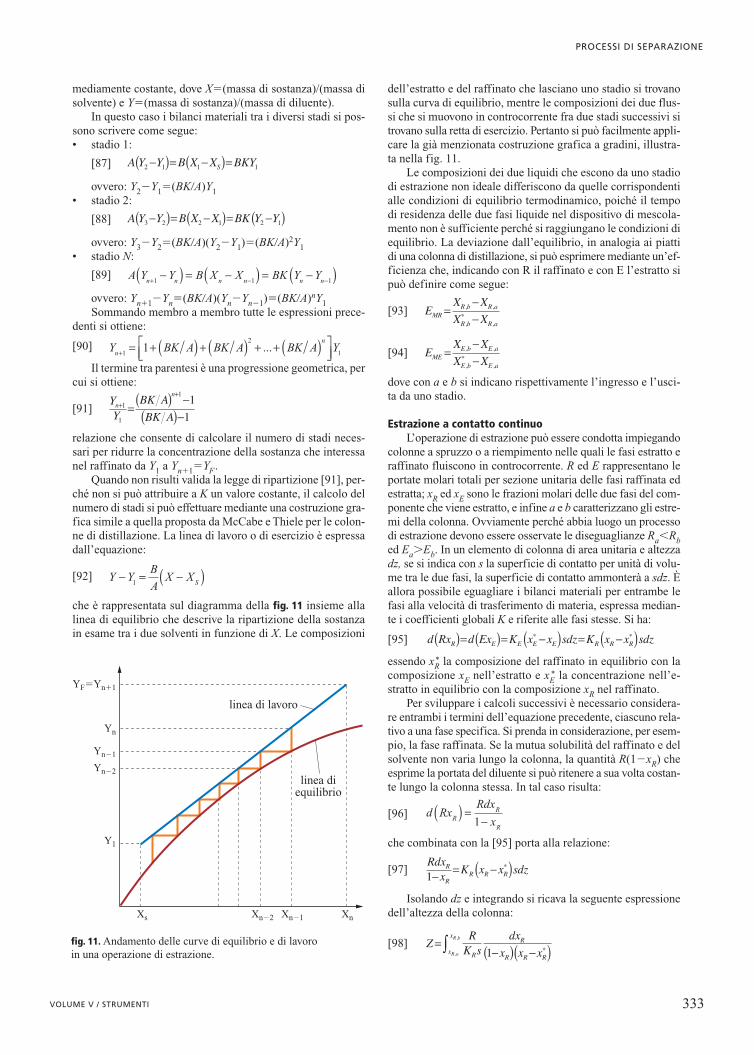
relazione che consente di calcolare il numero di stadi neces-sari per ridurre la concentrazione della sostanza che interessanel raffinato da Y1 a Yn�1�YF.
Quando non risulti valida la legge di ripartizione [91], per-ché non si può attribuire a K un valore costante, il calcolo delnumero di stadi si può effettuare mediante una costruzione gra-fica simile a quella proposta da McCabe e Thiele per le colon-ne di distillazione. La linea di lavoro o di esercizio è espressadall’equazione:
[92]
che è rappresentata sul diagramma della fig. 11 insieme allalinea di equilibrio che descrive la ripartizione della sostanzain esame tra i due solventi in funzione di X. Le composizioni
dell’estratto e del raffinato che lasciano uno stadio si trovanosulla curva di equilibrio, mentre le composizioni dei due flus-si che si muovono in controcorrente fra due stadi successivi sitrovano sulla retta di esercizio. Pertanto si può facilmente appli-care la già menzionata costruzione grafica a gradini, illustra-ta nella fig. 11.
Le composizioni dei due liquidi che escono da uno stadiodi estrazione non ideale differiscono da quelle corrispondentialle condizioni di equilibrio termodinamico, poiché il tempodi residenza delle due fasi liquide nel dispositivo di mescola-mento non è sufficiente perché si raggiungano le condizioni diequilibrio. La deviazione dall’equilibrio, in analogia ai piattidi una colonna di distillazione, si può esprimere mediante un’ef-ficienza che, indicando con R il raffinato e con E l’estratto sipuò definire come segue:
[93]
[94]
dove con a e b si indicano rispettivamente l’ingresso e l’usci-ta da uno stadio.
Estrazione a contatto continuoL’operazione di estrazione può essere condotta impiegando
colonne a spruzzo o a riempimento nelle quali le fasi estratto eraffinato fluiscono in controcorrente. R ed E rappresentano leportate molari totali per sezione unitaria delle fasi raffinata edestratta; xR ed xE sono le frazioni molari delle due fasi del com-ponente che viene estratto, e infine a e b caratterizzano gli estre-mi della colonna. Ovviamente perché abbia luogo un processodi estrazione devono essere osservate le diseguaglianze Ra�Rbed Ea�Eb. In un elemento di colonna di area unitaria e altezzadz, se si indica con s la superficie di contatto per unità di volu-me tra le due fasi, la superficie di contatto ammonterà a sdz. Èallora possibile eguagliare i bilanci materiali per entrambe lefasi alla velocità di trasferimento di materia, espressa median-te i coefficienti globali K e riferite alle fasi stesse. Si ha:
[95]
essendo xR* la composizione del raffinato in equilibrio con la
composizione xE nell’estratto e xE* la concentrazione nell’e-
stratto in equilibrio con la composizione xR nel raffinato. Per sviluppare i calcoli successivi è necessario considera-
re entrambi i termini dell’equazione precedente, ciascuno rela-tivo a una fase specifica. Si prenda in considerazione, per esem-pio, la fase raffinata. Se la mutua solubilità del raffinato e delsolvente non varia lungo la colonna, la quantità R(1�xR) cheesprime la portata del diluente si può ritenere a sua volta costan-te lungo la colonna stessa. In tal caso risulta:
[96]
che combinata con la [95] porta alla relazione:
[97]
Isolando dz e integrando si ricava la seguente espressionedell’altezza della colonna:
[98] Z RK s
dxx x xRx
xR
R R RR a
R b=−( ) −( )∫ ∗
,
,
1
Rdxx K x x sdzR
RR R R1− = −( )∗
d RxRdx
xRR
R
( ) = −1
d Rx d Ex K x x sdz K x x sdzR E E E E R R R( )= ( )= −( ) = −( )∗ ∗
EX XX XME
E b E a
E b E a
=−−∗
, ,
, ,
EX XX XMR
R b R a
R b R a
=−−∗
, ,
, ,
Y Y BA
X XS− = −( )1
YY
BK ABK A
nn
++
=( ) −( )−
1
1
1 11
Y BK A BK A BK A Yn
n
+ = + ( ) + ( ) + + ( )
1
2
11 ...
A Y Y B X X BK Y Yn n n n n n+ − −−( ) = −( ) = −( )1 1 1
A Y Y B X X BK Y Y3 2 2 1 2 1−( )= −( )= −( )
A Y Y B X X BKYS2 1 1 1−( )= −( )=
PROCESSI DI SEPARAZIONE
333VOLUME V / STRUMENTI
Xs Xn�2 Xn�1
Yn�1
Yn
Yn�2
Y1
YF�Yn�1
Xn
linea di lavoro
linea diequilibrio
fig. 11. Andamento delle curve di equilibrio e di lavoro in una operazione di estrazione.

In molti casi pratici xR non è molto elevato, e quindi è legit-timo porre (1�xR)�1. Se inoltre si assume che entrambi R eKR siano costanti lungo la colonna, l’equazione precedentediventa:
[99]
che può essere scritta nella forma
[100]
avendo definito, analogamente a quanto fatto nell’equazione[83],
[101]
detto ‘numero di unità di trasferimento’, mentre HOR�(R�KRs)viene detta ‘altezza di una unità di trasferimento’.
Il calcolo dell’integrale che compare nella [101] si può ese-guire valutando la differenza (xR�xR
*) da un diagramma simi-le a quello illustrato nella fig. 11, in cui vengono riportate siala curva di lavoro, che lega xR a xE in ogni punto della colon-na e ottenuta dal bilancio materiale, sia la curva di equilibrio,che lega le composizioni di xR e xE in condizioni di equilibriotermodinamico.
Si ricorda inoltre per inciso che, nel caso in cui il solven-te e il raffinato siano insolubili l’uno nell’altro, l’unico pro-cesso che ha luogo nella colonna è il trasferimento del com-ponente solubile dalla fase R alla fase E, per cui la linea dilavoro è espressa dalla relazione:
[102]
analoga alla [78] per la colonna di assorbimento. Rs ed Es espri-mono le portate del raffinato e del solvente puri, cioè liberi daaltri componenti.
6.2.6 Estrazione solido-liquido ed estrazione con solventi supercritici
L’estrazione di un componente solubile da un materiale solidomediante l’uso di un solvente trova le sue prime applicazioniin un’epoca che precede di molto la nascita dell’industria chi-mica, poiché fin dall’antichità tale tecnica veniva utilizzata perricavare sali alcalini dalle ceneri di legno.
Un metodo semplice ampiamente usato consiste nel rico-prire il solido con il solvente entro un contenitore di acciaio digrande capacità e lasciare il sistema in questo stato per un tempoprolungato. L’estrazione avviene di solito a temperatura ambien-te, e quindi senza rischi di decomposizione per i composti ter-molabili, ma i lunghi tempi richiesti sono dovuti alla lentezzadei processi diffusivi, soprattutto nel solido. L’innalzamentodella temperatura, con ovvi problemi per le sostanze termola-bili, e l’uso degli ultrasuoni possono essere usati per accelera-re il processo estrattivo. Per ridurre i tempi, se la quantità disolido da trattare è molto elevata, conviene caricare il solido inuna colonna di acciaio all’interno della quale viene fatto scor-rere il liquido estraente. Poiché le rese per passaggio non sonoelevate, tale liquido viene abitualmente fatto ricircolare piùvolte. In questo modo il tempo risulta notevolmente ridotto.
Una tecnica relativamente recente per estrarre i componen-ti presenti in una fase condensata utilizza gas che si trovano a
una temperatura al di sopra di quella critica, ovvero che si tro-vano in quella regione del diagramma di stato nella quale uncomposto, puro, non può essere liquefatto, qualunque sia lapressione applicata. Il punto critico si trova infatti al limitedella curva che definisce la zona bifasica nella quale coesi-stono il liquido e il vapore (v. cap. 2.3).
Un fluido supercritico ha proprietà intermedie tra quelleliquide e quelle gassose, possedendo le caratteristiche solven-ti dei liquidi e quelle di trasporto dei gas. Esso può quindi esse-re descritto come un liquido molto mobile. Ciò permette diottenere delle velocità e delle efficienze di estrazione moltomigliori di quelle che si ottengono nei processi convenziona-li. Inoltre le condizioni di estrazione possono essere variate inmodo tale da ottenere delle separazioni ben controllate. L’e-strazione con fluidi supercritici dipende dalla densità del flui-do, che può essere modificata variando pressione e tempera-tura del sistema. Il potere solvente di un fluido supercritico cre-sce a parità di temperatura all’aumentare della densità, o aparità di densità all’aumentare della temperatura. Un fluidosupercritico può essere utilizzato per estrarre un soluto da unsolido, con il vantaggio che, contrariamente a quanto avvienein un’estrazione convenzionale, quando le condizioni ridiven-tano quelle ambientali, il solvente, molto volatile, abbandonaquasi totalmente il materiale estratto.
Il principio fondamentale dell’estrazione con fluidi super-critici consiste nel fatto che la solubilità di un solvente variacon la pressione e la temperatura. In condizioni ambiente lasolubilità di un soluto in un gas è correlata alla tensione divapore del soluto ed è generalmente trascurabile, ma in un sol-vente supercritico le solubilità risultano di 10 ordini di gran-dezza superiori rispetto a quelle che si prevedono in base allalegge dei gas ideali.
L’estrazione supercritica non ha ancora trovato un ampioimpiego nell’industria, ma sta diventando sempre più attraen-te sia per le elevate purezze che consente di raggiungere siaper ragioni di impatto ambientale, dato che non si usano sol-venti tossici. Inoltre l’estrazione supercritica permette di otte-nere separazioni che con processi convenzionali sarebbero piut-tosto difficoltose, e opera a temperature generalmente basse,senza perciò dare problemi di decomposizione se si trattanocomposti termolabili. Gli svantaggi più significativi dell’e-strazione supercritica sono: la necessità di operare a una pres-sione elevata; la presenza di complesse operazioni di riciclag-gio per ridurre i costi energetici dovuti alla compressione delsolvente; gli elevati investimenti per le apparecchiature neces-sarie.
Il biossido di carbonio è il solvente più comune, soprat-tutto perché i valori dei suoi parametri critici sono piuttostobassi (31 °C; 74 bar); inoltre è economico e atossico. I solventiorganici, di impiego piuttosto ampio in petrolchimica, sonoinvece esplosivi e, di conseguenza, richiedono apparecchiatu-re con un costo molto elevato. I clorofluorocarburi avrebberoeccellenti proprietà come solventi supercritici, ma il loro impie-go è limitato a causa dei loro effetti dannosi sull’ozonosfera.
L’estrazione supercritica trova applicazioni nell’industriaalimentare, per esempio nelle operazioni di decaffeneizzazio-ne oppure nelle estrazioni di aromi e oli essenziali dalle spe-zie, e nell’industria farmaceutica, dove serve per ottenere i prin-cipi attivi dalle piante senza l’azione potenzialmente distrutti-va del calore ed evitando la presenza di solvente residuo nelprodotto. Nell’industria petrolchimica l’impiego più comunedell’estrazione supercritica è il trattamento del residuo di estra-zione del greggio.
Rxx
xx E
xx
xs
R a
R a
R
Rs
E a
E a
E,
,
,
,1 1 1 1− − −
= − − −xxE
N dxx xOR
R
R Rx
x
R a
R b=−( )∗∫
,
,
Z H NOR OR=
Z RK s
dxx xR
R
R Rx
x
R a
R b=−( )∗∫
,
,
ASPETTI PROCESSISTICI
334 ENCICLOPEDIA DEGLI IDROCARBURI
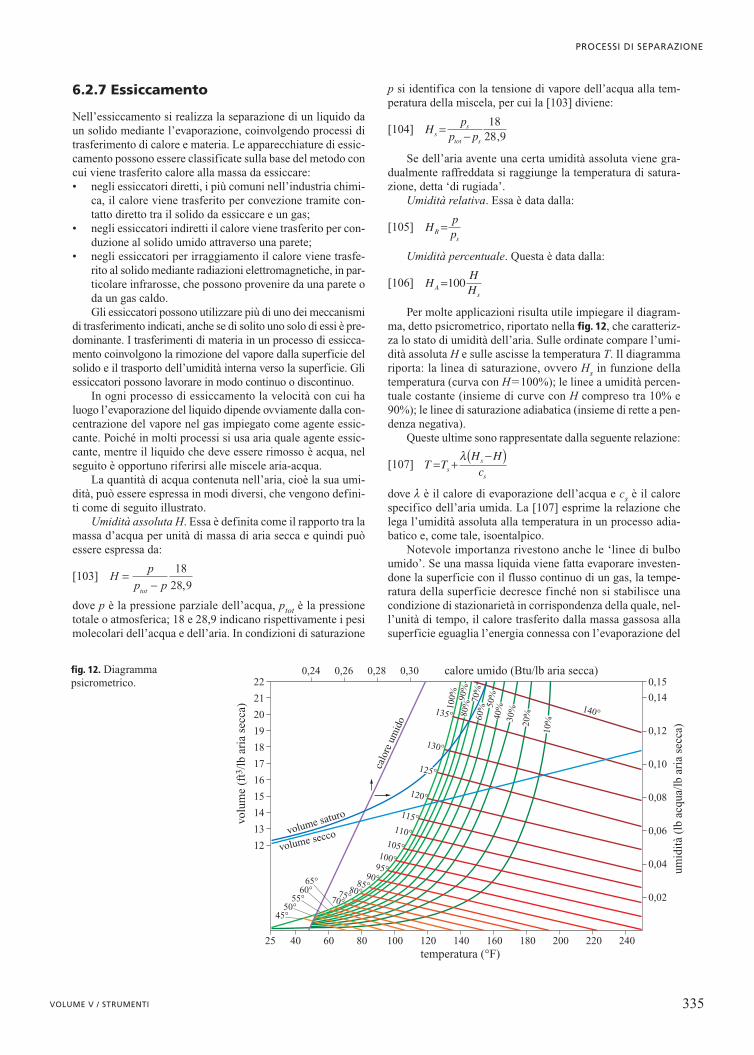
6.2.7 Essiccamento
Nell’essiccamento si realizza la separazione di un liquido daun solido mediante l’evaporazione, coinvolgendo processi ditrasferimento di calore e materia. Le apparecchiature di essic-camento possono essere classificate sulla base del metodo concui viene trasferito calore alla massa da essiccare:• negli essiccatori diretti, i più comuni nell’industria chimi-
ca, il calore viene trasferito per convezione tramite con-tatto diretto tra il solido da essiccare e un gas;
• negli essiccatori indiretti il calore viene trasferito per con-duzione al solido umido attraverso una parete;
• negli essiccatori per irraggiamento il calore viene trasfe-rito al solido mediante radiazioni elettromagnetiche, in par-ticolare infrarosse, che possono provenire da una parete oda un gas caldo.Gli essiccatori possono utilizzare più di uno dei meccanismi
di trasferimento indicati, anche se di solito uno solo di essi è pre-dominante. I trasferimenti di materia in un processo di essicca-mento coinvolgono la rimozione del vapore dalla superficie delsolido e il trasporto dell’umidità interna verso la superficie. Gliessiccatori possono lavorare in modo continuo o discontinuo.
In ogni processo di essiccamento la velocità con cui haluogo l’evaporazione del liquido dipende ovviamente dalla con-centrazione del vapore nel gas impiegato come agente essic-cante. Poiché in molti processi si usa aria quale agente essic-cante, mentre il liquido che deve essere rimosso è acqua, nelseguito è opportuno riferirsi alle miscele aria-acqua.
La quantità di acqua contenuta nell’aria, cioè la sua umi-dità, può essere espressa in modi diversi, che vengono defini-ti come di seguito illustrato.
Umidità assoluta H. Essa è definita come il rapporto tra lamassa d’acqua per unità di massa di aria secca e quindi puòessere espressa da:
[103]
dove p è la pressione parziale dell’acqua, ptot è la pressionetotale o atmosferica; 18 e 28,9 indicano rispettivamente i pesimolecolari dell’acqua e dell’aria. In condizioni di saturazione
p si identifica con la tensione di vapore dell’acqua alla tem-peratura della miscela, per cui la [103] diviene:
[104]
Se dell’aria avente una certa umidità assoluta viene gra-dualmente raffreddata si raggiunge la temperatura di satura-zione, detta ‘di rugiada’.
Umidità relativa. Essa è data dalla:
[105]
Umidità percentuale. Questa è data dalla:
[106]
Per molte applicazioni risulta utile impiegare il diagram-ma, detto psicrometrico, riportato nella fig. 12, che caratteriz-za lo stato di umidità dell’aria. Sulle ordinate compare l’umi-dità assoluta H e sulle ascisse la temperatura T. Il diagrammariporta: la linea di saturazione, ovvero Hs in funzione dellatemperatura (curva con H�100%); le linee a umidità percen-tuale costante (insieme di curve con H compreso tra 10% e90%); le linee di saturazione adiabatica (insieme di rette a pen-denza negativa).
Queste ultime sono rappresentate dalla seguente relazione:
[107]
dove l è il calore di evaporazione dell’acqua e cs è il calorespecifico dell’aria umida. La [107] esprime la relazione chelega l’umidità assoluta alla temperatura in un processo adia-batico e, come tale, isoentalpico.
Notevole importanza rivestono anche le ‘linee di bulboumido’. Se una massa liquida viene fatta evaporare investen-done la superficie con il flusso continuo di un gas, la tempe-ratura della superficie decresce finché non si stabilisce unacondizione di stazionarietà in corrispondenza della quale, nel-l’unità di tempo, il calore trasferito dalla massa gassosa allasuperficie eguaglia l’energia connessa con l’evaporazione del
T TH Hcss
s= +
−( )λ
H HHA
s=100
H ppRs
=
H pp ps
s
tot s= −
1828 9,
H pp ptot
=−
18
28 9,
PROCESSI DI SEPARAZIONE
335VOLUME V / STRUMENTI
volu
me
(ft3
/lb
aria
sec
ca)
umid
ità
(lb
acqu
a/lb
ari
a se
cca)
13
12
14
15
16
17
18
19
20
21
22
0,02
0,04
0,06
0,08
0,10
0,12
0,14
0,15
temperatura (°F)
calore umido (Btu/lb aria secca)
calo
re u
mid
o
volume saturo
volume secco
25 40
0,24 0,26 0,28 0,30
10%20
%30%
40%50
%
60%
70%
80%
90%
100%
60 80 100 120 140 160
135°140°
130°
125°
120°
115°110°
105°100°
95°90°85°80°75°70°
65°60°
55°50°
45°
180 200 220 240
fig. 12. Diagrammapsicrometrico.

liquido. La temperatura Ts della superficie liquida in tal casoè detta di bulbo umido, mentre la pressione parziale del flui-do evaporante in corrispondenza della superficie stessa ugua-glia la tensione di vapore del liquido alla temperatura Ts. Il pro-cesso descritto soddisfa l’equazione:
[108]
dove il primo membro rappresenta il calore trasmesso dal gasper convezione, mentre il secondo è l’energia connessa con l’e-vaporazione. In particolare, l è il calore molare di evapora-zione, h è il coefficiente di trasferimento di calore dal gas alliquido, kg è il coefficiente di trasporto di materia, T è la tem-peratura del gas, Ts è la temperatura della superficie liquida,ps è la tensione di vapore dell’acqua alla temperatura Ts, (ps�p)è la forza motrice del processo di evaporazione espressa comedifferenza tra la pressione parziale dell’acqua in corrispon-denza della superficie evaporante e la sua pressione parzialenell’aria.
Ricordando la [107], la [108] si può riscrivere:
[109]
dove kg��kg(28,9�18)p. Le linee che descrivono l’ultima rela-zione vengono appunto chiamate di bulbo umido. In realtà peril sistema aria-acqua si verifica che casualmente il rapportoh�kg� è molto prossimo a cs. Per tale ragione le linee di satura-zione adiabatica e le linee di bulbo umido nel diagramma difig. 12 sono coincidenti.
Le linee di bulbo umido sono utili per poter determinarel’umidità di una miscela gassosa. A tale scopo è necessariovalutare la temperatura della miscela (detta temperatura dibulbo secco) e quella ottenuta con un termometro a contattocon una garza imbevuta di liquido, su cui viene insufflato ilgas. L’umidità assoluta della miscela si può allora ricavare conil diagramma della fig. 12, tramite una semplice costruzionenella quale si individua il punto di intersezione della retta chedescrive la [109] e la curva di saturazione.
Per poter dimensionare e stabilire le capacità operative diun essiccatore è necessario conoscere la velocità con cui si svol-ge il processo di essiccamento. Essa infatti permette di ricava-re il tempo necessario per portare il solido al grado desideratodi umidità e quindi di stabilire il tempo di residenza del solidostesso nell’apparecchiatura. Se si eseguono esperimenti di essic-camento su un solido misurando il suo contenuto di liquido W(in kg/kg di solido secco) in funzione del tempo, si possonovalutare i valori della velocità di evaporazione (dW�dt)�(DW/Dt)in funzione del tempo. Si osserva che la velocità di evapora-zione si mantiene costante fino a un tempo critico, al di là delquale decresce con regolarità. Questo risultato può essere inter-pretato ipotizzando che durante il periodo iniziale il processodi evaporazione coinvolga soltanto la parte esterna del solido,e quindi si possa descrivere come un semplice processo di eva-porazione di un liquido che ricopre completamente la superfi-cie esterna del solido. Durante il periodo successivo, invece, ilprocesso coinvolge il liquido trattenuto all’interno delle parti-celle solide, e quindi l’evaporazione coinvolge processi più com-plessi. Nel periodo iniziale, la cinetica di evaporazione si iden-tifica con la velocità di trasferimento di materia dalla superfi-cie del solido al cuore della massa fluida essiccante. La cineticache descrive il processo si può esprimere con un’equazione incui, con riferimento all’unità di tempo e all’unità di volume disolido, si uguaglia l’energia connessa con l’evaporazione del-l’acqua con il calore trasmesso dal fluido essiccante:
Queste ultime sono rappresentate dalla seguente relazione:
[110]
dove Ts è la temperatura della superficie di evaporazione, rs ladensità del solido secco, a la superficie per volume unitariodel solido; l è riferito alla massa anziché alle moli. Dalla rela-zione precedente si ricava:
[111]
Dopo il punto critico, la velocità di essiccamento dipendedal moto del liquido all’interno del solido e, di conseguenza,l’influenza delle variabili esterne diviene di minore importan-za. Il processo di evaporazione risulta controllato dalla diffu-sione del liquido all’interno del solido e pertanto la velocità dievaporazione si deduce integrando l’equazione della diffusio-ne. Per uno strato di spessore l vale la relazione:
[112]
dove Dl è il coefficiente di diffusione nel liquido e We indicail valore dell’umidità nel solido, detta all’equilibrio, cui essopuò essere portato a contatto con un gas a una data umidità ea una determinata temperatura. Integrando la [112], si ottienela seguente espressione del tempo di essiccamento:
[113]
dove Wc indica il contenuto di liquido nel solido al punto cri-tico.
In un letto di particelle solide di elevata porosità, un liqui-do può muoversi da una regione a un’altra sotto l’effetto diforze capillari. Infatti, quando il liquido viene rimosso pro-gressivamente dal letto, la curvatura delle superfici liquidenelle parti esterne degli interstizi porosi aumenta, dando luogoa una pressione che tende a risucchiare il liquido dall’internoverso la superficie esterna. All’avanzare del processo di essic-camento, la curvatura del menisco aumenta, finché la forza di‘risucchio’ raggiunge un valore tale da provocare la rottura delporo in superficie; entra quindi aria e l’umidità interna del soli-do si ridistribuisce a un livello più basso.
La velocità di essiccamento nei processi che non siano con-dizionati dalla diffusione può essere espressa mediante la rela-zione:
[114]
La costante Kl può essere valutata applicando in corri-spondenza del punto critico di essiccamento la relazione
[115]
Se si sostituisce nella [115] il valore di (dW�dt)c ricavatodalla [111], si ottiene:
[116]
Integrando la relazione così ottenuta si ricava per il tempodi essiccamento:
[117] tW W
ha T TW WW W
s c e
s
e
e
=−( )−( )
−−
rλln
0
Kha T T
W Wls
s c e
= −−( )−( )r λ
KdW dtW Wl
c
c e
= −( )
−
dWdt
K W Wl e= − −( )
t lD
W WW W
l
c e
e
=−−
4 2
2πln
dWdt
Dl
W Wle= −( )π2
24
dWdt
ha T Ts
s= −( )r λ
rs sdWdt ah T Tλ = −( )
H H hk
T Tsg
s− = −( )�λ
h T T k p ps g s−( )= −( )λ
ASPETTI PROCESSISTICI
336 ENCICLOPEDIA DEGLI IDROCARBURI
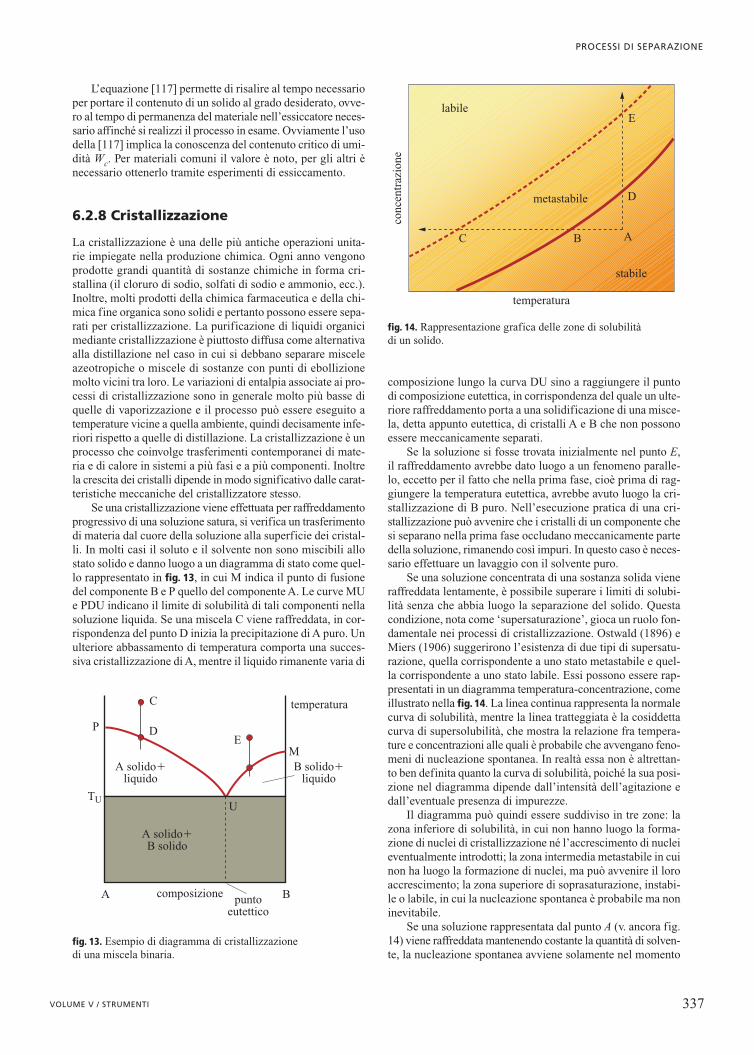
L’equazione [117] permette di risalire al tempo necessarioper portare il contenuto di un solido al grado desiderato, ovve-ro al tempo di permanenza del materiale nell’essiccatore neces-sario affinché si realizzi il processo in esame. Ovviamente l’usodella [117] implica la conoscenza del contenuto critico di umi-dità Wc. Per materiali comuni il valore è noto, per gli altri ènecessario ottenerlo tramite esperimenti di essiccamento.
6.2.8 Cristallizzazione
La cristallizzazione è una delle più antiche operazioni unita-rie impiegate nella produzione chimica. Ogni anno vengonoprodotte grandi quantità di sostanze chimiche in forma cri-stallina (il cloruro di sodio, solfati di sodio e ammonio, ecc.).Inoltre, molti prodotti della chimica farmaceutica e della chi-mica fine organica sono solidi e pertanto possono essere sepa-rati per cristallizzazione. La purificazione di liquidi organicimediante cristallizzazione è piuttosto diffusa come alternativaalla distillazione nel caso in cui si debbano separare misceleazeotropiche o miscele di sostanze con punti di ebollizionemolto vicini tra loro. Le variazioni di entalpia associate ai pro-cessi di cristallizzazione sono in generale molto più basse diquelle di vaporizzazione e il processo può essere eseguito atemperature vicine a quella ambiente, quindi decisamente infe-riori rispetto a quelle di distillazione. La cristallizzazione è unprocesso che coinvolge trasferimenti contemporanei di mate-ria e di calore in sistemi a più fasi e a più componenti. Inoltrela crescita dei cristalli dipende in modo significativo dalle carat-teristiche meccaniche del cristallizzatore stesso.
Se una cristallizzazione viene effettuata per raffreddamentoprogressivo di una soluzione satura, si verifica un trasferimentodi materia dal cuore della soluzione alla superficie dei cristal-li. In molti casi il soluto e il solvente non sono miscibili allostato solido e danno luogo a un diagramma di stato come quel-lo rappresentato in fig. 13, in cui M indica il punto di fusionedel componente B e P quello del componente A. Le curve MUe PDU indicano il limite di solubilità di tali componenti nellasoluzione liquida. Se una miscela C viene raffreddata, in cor-rispondenza del punto D inizia la precipitazione di A puro. Unulteriore abbassamento di temperatura comporta una succes-siva cristallizzazione di A, mentre il liquido rimanente varia di
composizione lungo la curva DU sino a raggiungere il puntodi composizione eutettica, in corrispondenza del quale un ulte-riore raffreddamento porta a una solidificazione di una misce-la, detta appunto eutettica, di cristalli A e B che non possonoessere meccanicamente separati.
Se la soluzione si fosse trovata inizialmente nel punto E,il raffreddamento avrebbe dato luogo a un fenomeno paralle-lo, eccetto per il fatto che nella prima fase, cioè prima di rag-giungere la temperatura eutettica, avrebbe avuto luogo la cri-stallizzazione di B puro. Nell’esecuzione pratica di una cri-stallizzazione può avvenire che i cristalli di un componente chesi separano nella prima fase occludano meccanicamente partedella soluzione, rimanendo così impuri. In questo caso è neces-sario effettuare un lavaggio con il solvente puro.
Se una soluzione concentrata di una sostanza solida vieneraffreddata lentamente, è possibile superare i limiti di solubi-lità senza che abbia luogo la separazione del solido. Questacondizione, nota come ‘supersaturazione’, gioca un ruolo fon-damentale nei processi di cristallizzazione. Ostwald (1896) eMiers (1906) suggerirono l’esistenza di due tipi di supersatu-razione, quella corrispondente a uno stato metastabile e quel-la corrispondente a uno stato labile. Essi possono essere rap-presentati in un diagramma temperatura-concentrazione, comeillustrato nella fig. 14. La linea continua rappresenta la normalecurva di solubilità, mentre la linea tratteggiata è la cosiddettacurva di supersolubilità, che mostra la relazione fra tempera-ture e concentrazioni alle quali è probabile che avvengano feno-meni di nucleazione spontanea. In realtà essa non è altrettan-to ben definita quanto la curva di solubilità, poiché la sua posi-zione nel diagramma dipende dall’intensità dell’agitazione edall’eventuale presenza di impurezze.
Il diagramma può quindi essere suddiviso in tre zone: lazona inferiore di solubilità, in cui non hanno luogo la forma-zione di nuclei di cristallizzazione né l’accrescimento di nucleieventualmente introdotti; la zona intermedia metastabile in cuinon ha luogo la formazione di nuclei, ma può avvenire il loroaccrescimento; la zona superiore di soprasaturazione, instabi-le o labile, in cui la nucleazione spontanea è probabile ma noninevitabile.
Se una soluzione rappresentata dal punto A (v. ancora fig.14) viene raffreddata mantenendo costante la quantità di solven-te, la nucleazione spontanea avviene solamente nel momento
PROCESSI DI SEPARAZIONE
337VOLUME V / STRUMENTI
temperatura
puntoeutettico
A solido�liquido
composizione
A solido�B solido
U
C
EM
P
TU
A B
D
B solido�liquido
fig. 13. Esempio di diagramma di cristallizzazione di una miscela binaria.
conc
entr
azio
ne
temperatura
stabile
metastabile
labileE
D
ABC
fig. 14. Rappresentazione grafica delle zone di solubilità di un solido.

in cui si raggiungono le condizioni rappresentate dal punto C.La tendenza alla nucleazione aumenta via via che si entra nellaregione soprasatura, a meno che la soluzione diventi così visco-sa da prevenirla. D’altra parte è possibile raggiungere le con-dizioni di soprasaturazione anche evaporando solvente dallasoluzione. La linea ADE rappresenta un’operazione di questotipo, condotta a temperatura costante. Operando in questo modoè però difficile entrare nella regione di soprasaturazione, per-ché la parte della soluzione che si trova vicina alla superficiedi evaporazione risulta più soprasatura di quella presente nelcuore della soluzione, per cui i cristalli generati alla superfi-cie penetrano nella soluzione, inducendo altri fenomeni dinucleazione, anche prima che il cuore della soluzione abbiaraggiunto le condizioni rappresentate dal punto E.
In ogni caso la nucleazione rappresenta un cambiamentoda una situazione termodinamicamente instabile a una meta-stabile. Il processo può essere schematizzato in questo modo:fase omogenea A��nuclei cristallini dispersi in A.
Esso ha luogo spontaneamente ed è associato a una dimi-nuzione dell’energia libera di Gibbs, che può essere a sua voltascissa nella somma di due termini, il primo dei quali, positivo,rappresenta il lavoro di formazione della superficie tra le duefasi, mentre il secondo, negativo, rappresenta la diminuzionedi energia libera associata alla formazione della nuova fase. Sipuò ricavare la seguente espressione della variazione globaledi energia libera:
[118]
dove s è la tensione interfacciale, r il raggio del nucleo (assun-to sferico) e rc il suo valore critico, che deve essere raggiuntoaffinché la nucleazione possa procedere spontaneamente. Sipuò facilmente dimostrare che a rc corrisponde un massimo diDG, cioè uno stato instabile in corrispondenza del quale unaparticella ha uguale probabilità di crescere o decrescere didimensioni, poiché in entrambi i casi si verifica una diminu-zione di energia libera.
Dal punto di vista cinetico il fenomeno della cristallizza-zione presenta due tipiche situazioni: la prima si riferisce allavelocità di formazione dei nuclei nella zona di soprasatura-zione, mentre la seconda alla loro velocità di accrescimento inentrambe le regioni, soprasatura e metastabile.
La formazione dei nuclei costituisce un tipico processoattivato la cui velocità, Rn (numero di nuclei formati/tempo),è data da:
[119]
dove W è il lavoro isotermo di formazione di un nucleo, cherisulta infinito in corrispondenza della curva di soprasatura-zione e che diminuisce all’aumentare della soprasaturazionestessa. Ne consegue che, aumentando il grado di soprasatura-zione, aumenta esponenzialmente la formazione di un nume-ro elevato di piccoli cristalli. Quindi, se si desidera la forma-zione di cristalli di grandi dimensioni, bisogna operare a bassasoprasaturazione, in modo tale che il processo proceda essen-zialmente per accrescimento.
La velocità di accrescimento in condizioni stazionarie sipuò esprimere come:
[120]
dove m è la massa di solido che viene depositata, D il coefficien-te di diffusione del componente che cristallizza in soluzione,
A la superficie del cristallo, d lo spessore dello strato liquidosuperficiale in cui è localizzata la resistenza alla diffusione.Con k si indica la costante di velocità del processo superficia-le di inserimento di uno ione sulla superficie del reticolo. Infi-ne C, Ci� e Cs rappresentano rispettivamente le concentrazionidel soluto nel cuore della soluzione, all’interfaccia e alla satu-razione. La [120] esprime quindi l’uguaglianza tra la velocitàdi trasporto del materiale alla superficie delle particelle soli-de e quella di formazione dello strato cristallino. EliminandoCi� la [120] può essere riscritta
[121]
dove (D�d)�kc esprime un coefficiente di trasporto di mate-ria. Il suo valore è condizionato da quello dello spessore dellostrato limite superficiale, che a sua volta dipende dalle condi-zioni fluidodinamiche in cui ha luogo il processo di cristalliz-zazione. Esistono in letteratura diverse relazioni espresse attra-verso l’impiego di parametri adimensionali che permettono divalutare tale coefficiente per particolari apparecchiature inopportune condizioni fisiche. In pratica si verifica che spessoil valore di kc è elevato rispetto a quello di k, per cui la cineti-ca globale del processo di cristallizzazione è controllata dallavelocità di accrescimento dei cristalli.
In ogni caso la simulazione di un processo di cristallizza-zione dipende dalla formulazione di un modello fisico che per-metta di descrivere e combinare insieme processi di nuclea-zione e crescita, in modo da poter determinare non solo l’am-montare di sostanza cristallizzata, ma anche la distribuzionedei cristalli aventi diverse dimensioni. Un modello generalepiuttosto utile è quello di Randolph e Larson (1962). Se si indi-ca con N il numero totale di cristalli contenuti in un determi-nato volume aventi una lunghezza caratteristica L, si può defi-nire una grandezza n, detta ‘densità di popolazione’:
[122]
Il prodotto n∆L esprime il numero di cristalli per volumeunitario aventi una lunghezza compresa in un piccolo inter-vallo ∆L�L1�L2. I suoi valori possono essere determinati spe-rimentalmente mediante un processo di classificazione mec-canica o per analisi ottica e microscopica nel caso di cristallidi piccole dimensioni.
Facendo poi riferimento a un cristallizzatore continuo divolume V ed effettuando un bilancio riferito alle particelle aven-ti un particolare valore di L, si ottiene, in condizioni staziona-rie, l’equazione:
[123]
essendo v la portata volumetrica del flusso uscente. Il primotermine dell’equazione precedente rappresenta l’aumentodel numero di particelle di lunghezza L all’interno del siste-ma, il secondo il loro trasporto verso l’esterno. In partico-lare, r è la velocità di produzione delle particelle di dime-nioni L, che esprime il numero di particelle di dimensioni Lche si producono per unità di tempo e unità di volume e sipuò riscrivere nella forma:
[124]
dove G�dL�dt è la velocità di crescita dei cristalli, poiché espri-me l’aumento della loro dimensione lineare L al variare del
r dndL
dLdt
dndLG=− =−
rV nv− =0
n NL
dNdLL
= =→
lim∆
∆∆0
dmdt
A C C
k D
s=−( )+1 δ
dmdt
D A C C kA C Ci i s= −( ) = −( )δ� �
R enW k TB∝ −
∆G r rrc
= −
4
2
3
23
πσ
ASPETTI PROCESSISTICI
338 ENCICLOPEDIA DEGLI IDROCARBURI

tempo. Il segno negativo presente nella precedente relazioneriflette il fatto che la variazione della densità di popolazione sideve esprimere come differenza fra il numero di particelle cheentrano nella dimensione L e quelle che ne escono, entrambeper effetto della crescita. Si può ora assumere che il valore diG per un insieme di cristalli geometricamente simili, dello stes-so materiale, sospesi nella stessa soluzione, sia uguale per tuttii cristalli, indipendentemente dalle loro dimensioni iniziali.Questa proposta dovuta a McCabe, è nota come ‘legge deltaL’ e si è rivelata valida per molti materiali con cristalli aventidimensioni inferiori a 50 mesh.
Applicandola alla [123] si ricava
[125]
avendo indicato con ÿ�V�v il tempo di residenza medio dellasospensione nel cristallizzatore. Se si indica con n0 la den-sità di popolazione delle particelle aventi le dimensioni deinuclei di cristallizzazione, integrando l’equazione preceden-te si ricava:
[126]
da cui:
[127]
in base alla quale esiste una relazione lineare tra lnn e L. Diver-si sistemi soddisfano la relazione [127]; tra essi si possono cita-re il solfato di potassio, lo zucchero, il cloruro di sodio, ilCaSO4�0,5H2O e l’urea. In altri casi la relazione tra lnn ed Lnon è lineare ed è necessario applicare modelli più sofisticatiper descrivere il fenomeno in esame.
L’utilità dell’equazione [127] risiede nel fatto che, median-te esperimenti di cristallizzazione condotti su piccola scala, èpossibile valutare i parametri del modello, n0 e G, in modo dapoter utilizzare i risultati ottenuti per dimensionare apparec-chiature di dimensioni maggiori.
Come menzionato, la cristallizzazione può essere ottenu-ta per refrigerazione o per evaporazione e i cristallizzatori si
dividono in due categorie che riflettono il processo seguito. Lafig. 15 illustra lo schema di un’unità di cristallizzazione perrefrigerazione, nella quale l’alimentazione al cristallizzatoreviene fatta fluire attraverso un refrigerante e quindi torna alrecipiente principale in cui ha luogo la cristallizzazione peraccrescimento. La circolazione del liquido, attuata tramite lapompa, contribuisce a tenere in agitazione uniforme la misce-la presente nel recipiente principale.
La cristallizzazione per evaporazione invece viene con-dotta in unità, come quella illustrata nella fig. 16, nelle quali ècontemplato che l’alimentazione venga preriscaldata prima diessere pompata nell’evaporatore per la rimozione parziale delsolvente. I cristalli vengono attinti alla base del recipiente, men-tre alla sommità del recipiente stesso si opera uno spurgo dellasoluzione concentrata.
Nelle operazioni condotte su larga scala il calore vienefornito mediante condensazione di vapore. Poiché la mag-gior parte dell’energia richiesta da un evaporatore è quellache corrisponde alla rimozione del solvente, è opportunorecuperare il contenuto termico del vapore prodotto. Ciò puòessere ottenuto mediante impianti a effetto multiplo, in cuiil vapore prodotto nel primo evaporatore viene impiegatocome agente di riscaldamento nel secondo evaporatore, ecosì via. Affinché ciò sia possibile, è necessario che la pres-sione di evaporazione diminuisca nel passaggio da un eva-poratore a quello successivo, e quindi l’ultimo evaporatoredella serie viene tenuto sotto vuoto mediante un dispositivobarometrico.
n n e L G= −0 ϑ
dnn
dLGn
n L
0 0∫ ∫=− ϑ
dndL
nVG
nG=− =−ν
ϑ
PROCESSI DI SEPARAZIONE
339VOLUME V / STRUMENTI
uscitacristalli
refrigerante
liquidomadre
alimentazione
pompacircolazione
fig. 15. Schema operativo di un cristallizzatore con refrigerazione.
acqua diraffreddamento
vapore
scam
biat
ore
uscitacondensato
gas noncondensabili
prodotto
cond
ensa
tore
baro
met
rico
alimentazione
fig. 16. Cristallizzazione per evaporazione a circolazione forzata.

6.2.9 Adsorbimento
L’adsorbimento è il fenomeno che permette l’accumulo selet-tivo su una superficie solida di particolari tipi di molecole con-tenute in un fluido a contatto con la superficie stessa. In altritermini, esso riguarda il processo di ripartizione di diversi com-ponenti in corrispondenza delle interfasi solido-fluido e si dif-ferenzia dal fenomeno dell’assorbimento nel quale ha luogo ladistribuzione dei componenti tra il cuore di due fasi, una liqui-da e una gassosa. Solitamente i solidi adsorbenti sono costi-tuiti da particelle porose granulari con elevate superfici com-prese tra 102 e 103 m2/g. La loro capacità adsorbente può supe-rare il 20% della massa del solido stesso.
Industrialmente l’adsorbimento è un metodo di separazio-ne che trova applicazione in diversi tipi di processo, tra i qualimeritano di essere citati: a) la decolorazione di liquidi e di fra-zioni petrolifere; b) la deodorazione di acque; c) la rimozionedi sostanze nocive da gas; d ) il recupero di vitamine o altricomponenti da miscele di fermentazione; e) il recupero di sol-venti da miscele gassose; f ) la separazione di componenti pre-senti in miscele, nel caso in cui risulti difficile o al limite impos-sibile effettuarne una separazione per distillazione (per esem-pio la separazione degli xileni isomeri e il recupero di olefineleggere da gas di cracking).
Le applicazioni industriali in cui vengono utilizzati letti diparticelle adsorbenti sono numerose e per classificarle è oppor-tuno considerare sia la fase su cui viene condotta l’operazio-ne (gas o liquido) sia lo scopo cui essa è orientata (purifica-zione o separazione). Infatti l’adsorbimento viene impiegatoanche per la rimozione di prodotti indesiderati dalla correntedi un processo o da scarichi inquinanti. In questo caso la cor-rente fluida viene fatta fluire su un letto di particelle, ponen-do particolare attenzione al punto di sfondamento dell’impu-rezza, cioè all’istante in cui la sua concentrazione in uscitadalla colonna adsorbente supera il valore tollerabile.
La separazione per adsorbimento sfrutta invece la seletti-vità che particolari solidi manifestano rispetto ai componentipresenti in miscele fluide. Infatti, facendo fluire queste misce-le in una colonna contenente un letto fisso di particelle adsor-benti, si constata che i diversi componenti escono con tempidiversi, dipendenti dal loro grado di adsorbibilità. Un esempiodi adsorbimento selettivo è quello che si effettua in laborato-rio quando si opera un’analisi gascromatografica.
La tab. 3 riporta alcuni tipici materiali solidi adsorbenti,indicando anche alcune loro caratteristiche peculiari quali lasuperficie e la porosità. Tra di essi occupano un posto di par-ticolare importanza le zeoliti. La loro struttura è infatti carat-terizzata da cavità con caratteristiche geometriche ben definite,
in cui possono essere ospitate selettivamente le molecole chea loro volta, grazie alla loro configurazione geometrica, pre-sentano una corrispondenza morfologica con la cavità.
L’impiego su grande scala dei processi di separazione peradsorbimento è stato a lungo limitato dalle difficoltà di pro-gettazione e gestione delle unità in essi implicate, poiché èrichiesta una profonda conoscenza sia degli aspetti chimico-fisici dei fenomeni coinvolti, sia di quelli modellistici neces-sari per la loro simulazione. Infatti i più comuni procedimen-ti di separazione per adsorbimento coinvolgono l’impiego diletti fissi di particelle che operano in condizioni transitorie,alternando uno stadio di adsorbimento con uno di rigenera-zione. La situazione può essere illustrata considerando unadisposizione in cui sono presenti tre colonne. Quando le colon-ne A e B operano in adsorbimento, la colonna C si trova in rige-nerazione. Successivamente la colonna A non viene più ali-mentata ed è sottoposta a rigenerazione per trattamento con undesorbente o per riscaldamento, mentre l’alimentazione vienefatta fluire attraverso le colonne B e C. Nella fase seguente lacolonna B viene posta in rigenerazione, mentre l’alimentazio-ne fluisce attraverso le colonne C e A. Infine viene ripristina-ta la configurazione iniziale. Ovviamente un processo di que-sto tipo può essere condotto utilizzando anche un numero dicolonne superiore rispetto a quello preso in considerazione, eper poterlo gestire è necessario essere in grado di simulare inmaniera adeguata il comportamento transitorio di ciascunacolonna. Si conferma quindi l’opportunità di disporre di model-li che permettano di valutare come varia nel tempo la compo-sizione della corrente uscente da una colonna, ad alimenta-zione assegnata, e la concentrazione dei diversi componentiadsorbiti sul solido nei diversi punti della colonna stessa.
Le difficoltà ora menzionate possono essere superate se sieffettua la separazione in modo continuo, in controcorrente trafluido e solido. Un esempio lo si riscontra nel processo chia-mato Hypersorption, mediante il quale si separa l’etilene dalmetano. La fig. 17 illustra uno schema dell’impianto che èsostanzialmente costituito da una colonna di frazionamento,posizionata sopra una colonna di desorbimento, o stripping, avapore. L’alimentazione gassosa (costituita da etilene e meta-no) viene inserita vicino al centro del frazionatore e sale versol’alto con adsorbimento dell’etilene e di parte del metano. Unriflusso di etilene viene alimentato alla base per far desorbireil metano dall’adsorbente, soprattutto nella parte inferiore dellacolonna. Il vapor d’acqua fa desorbire a sua volta l’etilene dal-l’adsorbente, mentre l’umidità viene rimossa dal solido median-te un lavaggio con metano. Il movimento discendente del soli-do rappresenta il punto debole del processo ora descritto, inquanto comporta costose operazioni meccaniche; inoltre le
ASPETTI PROCESSISTICI
340 ENCICLOPEDIA DEGLI IDROCARBURI
tab. 3. Materiali impiegati nei processi di separazione per adsorbimento
Materiale Composizione chimicaArea superficiale
(m2/g)Porosità
Densitàapparente
Zeoliti Y Na12[(AlO2)12(SiO2)12]�27H2O (faujasiti) 824 0,45 1,1
Zeoliti X Na86[(AlO2)86(SiO2)106]�H2O (faujasiti) 410 0,57 1,18
Silica-gel SiO2 515 0,46 1,09
Allumina attivata Al2O3 354 0,61 1,25
Carbone attivato C 700-1.200 0,56 0,76

particelle solide sono soggette a sgretolamento e a contami-nazioni. Per questo motivo i letti mobili hanno ricevuto scar-se applicazioni, mentre è stato preferito l’uso di letti fissi.
Un’importante alternativa al processo precedente, che rap-presenta un compromesso fra l’impiego di un letto fisso e larealizzazione di un processo continuo, è l’operazione cosid-detta a letto mobile simulato (Broughton et al., 1970), che èstata applicata con successo nella separazione di miscele liqui-de di idrocarburi isomeri (in particolare gli xileni) e che con-siste nel tenere il letto adsorbente fisso, variando periodica-mente nel tempo le posizioni di alimentazione e rimozione delliquido.
Questo approccio viene realizzato industrialmente comeillustrato nella fig. 18, in cui una pompa fa circolare il liquidodal basso verso l’alto esternamente alla colonna. Un disposi-tivo, indicato con RV (Rotating Valve), valvola rotante, operain modo tale da dirigere ciascuna delle diverse correnti allevarie linee collegate con la colonna di adsorbimento. In ogni
momento solo quattro linee sono attive; nel caso della figurasono le linee 2, 5, 9 e 12. Questa configurazione permette diattuare contemporaneamente le operazioni di rigenerazione,nella parte superiore della colonna, e di adsorbimento, nellaparte inferiore. Quando l’elemento rotante della valvola simuove verso la posizione successiva, ciascun flusso viene tra-sferito alla linea adiacente. In questo modo il desorbente entranella linea 3 invece che nella 2, l’estratto viene prelevato dalla6 invece che dalla 5, l’alimentazione entra dalla 10 invece chedalla 9 e il raffinato viene sottratto dalla 1 invece che dalla 12.Le operazioni di adsorbimento e rigenerazione seguono quin-di l’evoluzione nel tempo delle concentrazioni dei diversi com-ponenti sul letto solido. Si tratta di un dispositivo ingegnoso,la cui attuazione risulta però costosa, tanto da limitarne l’usoa impianti di elevate produzioni, come quello già menzionatodella separazione degli xileni.
Di solito i letti adsorbenti su piccola e media scala vengo-no utilizzati operando in maniera semicontinua. Sono statianche realizzati impianti molto più semplici che sfruttano unatecnica di alimentazione a impulsi, cioè una quantità finita dimiscela adsorbibile in una corrente di desorbente (Seko et al.,1979). In questo caso l’andamento nel tempo di due compo-nenti A e B ha il tipico movimento a impulsi della cromato-grafia. Tramite opportuni prelievi essi possono essere separa-ti e, sfruttando più colonne, è possibile rendere continua taleoperazione. In questo modo il processo somiglia molto a unacromatografia di laboratorio. Le descrizioni precedenti mostra-no come l’applicazione industriale dei metodi di separazioneper adsorbimento richieda un’adeguata padronanza di model-li matematici che permettano di simulare il comportamentotransitorio delle unità. Nella formulazione di tali modelli èopportuno identificare i seguenti punti fondamentali: • studio dell’equilibrio termodinamico fluido-solido;• formulazione delle espressioni cinetiche che esprimono la
velocità di adsorbimento sulle singole particelle;• formulazione dei bilanci materiali delle colonne di adsor-
bimento e individuazione dei metodi di risoluzione delleequazioni così ottenute;
• analisi del comportamento delle unità di operazione in fun-zione delle variabili operative, quali la composizione e laportata dell’alimentazione, la temperatura di esercizio ecosì via.
Equilibri di adsorbimentoL’equilibrio di adsorbimento può essere descritto median-
te opportune isoterme che esprimono, a temperatura assegna-ta, la relazione esistente tra le pressioni parziali e la concen-trazione dei diversi componenti presenti nella fase fluida e quel-li esistenti sulla superficie del solido. Nei processi di separazioneinterviene soltanto l’adsorbimento di tipo fisico, provocato daforze di van der Waals, senza formazione di legami chimici.Lo studio di questi fenomeni peraltro risulta piuttosto compli-cato soprattutto quando il materiale adsorbente è dotato di unastruttura complessa, come si verifica nel caso delle zeoliti. L’e-quazione più usata per descrivere le isoterme di adsorbimen-to è quella di Langmuir:
[128]
dove gli indici i e j si riferiscono ai diversi componenti dellamiscela; qi è una variabile adimensionale che esprime il gradodi ricoprimento della superficie da parte del componente i,
θii i
j jj
nbC
b C=
+=∑1
1
PROCESSI DI SEPARAZIONE
341VOLUME V / STRUMENTI
CH4
C2H4
R
C
C
CH4�C2H4
C2H4
H2O
H2O
CH4
trasportosolido
vapore
fig. 17. Schema dell’impianto Hypersorption per la separazionedell’etilene dal metano. C, condensatore; R, vaporizzatore.
1
234567
R
F
F
E
D
89101112
R R
D
E
RVE
fig. 18. Processo a letto mobile simulato. E, estratto; R, raffinato; D, desorbente; F, alimentazione.

mentre Ci è la concentrazione di i nella fase fluida; i bi sonoopportuni parametri, detti costanti di adsorbimento, che soli-tamente vengono valutati per via sperimentale. Il grado di rico-primento viene espresso attraverso il rapporto qi�Gi �Gi
, doveGi è la concentrazione di i nella fase solida, mentre Gi
rappre-senta la capacità di carico dell’adsorbente. Il largo impiego del-l’equazione di Langmuir è dovuto al fatto che essa è in gradodi descrivere il raggiungimento delle condizioni di saturazio-ne del solido e di tener conto della composizione della misce-la fluida. Questo effetto è infatti importante per condizionarela selettività di un adsorbente nel riguardo dei vari componentiin gioco. L’isoterma di Fowler rappresenta un miglioramentorispetto a quella di Langmuir, poiché tiene conto della varia-zione del calore di adsorbimento con il grado di ricoprimento,dovuto all’eterogeneità della superficie e all’interazione tra lemolecole adsorbite (ci):
[129]
Questa equazione permette quindi di tenere conto dell’in-fluenza del grado di ricoprimento sulla selettività.
Cinetica dei processi di adsorbimentoPer quanto concerne gli aspetti cinetici bisogna tenere pre-
sente che l’adsorbimento è un tipico processo di trasferimen-to di materia che ha luogo attraverso la seguente successionedi stadi:• stadio 1: trasferimento esterno di materia, cioè dal cuore
del fluido alla superficie della particella; • stadio 2: diffusione nei pori della particella; • stadio 3: adsorbimento superficiale; • stadio 4: diffusione nel solido.
Questa situazione potrebbe riferirsi per esempio a un mate-riale come le zeoliti, in cui le particelle sono costituite da pic-coli microcristalli (1-3 mm), aggregati tra loro. In questo modoè possibile identificare una struttura porosa bimodale dove sonopresenti le microporosità del cristallo, in cui ha luogo lo sta-dio diffusivo 4, e la macroporosità intercristallina, in cui haluogo lo stadio diffusivo 2. Inoltre all’interno delle particellesi creano dei gradienti di concentrazione che ovviamente influen-zano la velocità globale del processo. Nel calcolo delle velo-cità di adsorbimento è possibile considerare alcune ipotesi sem-plificative:• l’influenza dello stadio 4 di diffusione interna si può tra-
scurare, se il rapporto tra diametro della particella e dia-metro dei microcristalli è elevato (come succede in moltezeoliti);
• lo stadio 3 può essere considerato all’equilibrio;• si possono, nel calcolo, utilizzare valori medi delle con-
centrazioni interne alle particelle, C_
i, evitando il gravosocalcolo dei gradienti di concentrazione.Sotto queste ipotesi il modello cinetico della singola par-
ticella si riduce a tre sole equazioni. La prima,
[130]
esprime l’impoverimento della fase fluida esterna per effettodel trasferimento di materia dal cuore del fluido alle particel-le, processo la cui velocità è proporzionale alla differenza trala concentrazione esterna del componente e quella media nellaparticella tramite un coefficiente globale Ki, che riflette la pre-senza di due resistenze in serie 1�Ki�1�ke�1�ki, dove ke è il
coefficiente di trasferimento esterno e ki quello interno. Il coef-ficiente esterno si può valutare dalle caratteristiche fluidodi-namiche del sistema, mentre quello interno dipende dal coef-ficiente di diffusione Di del componente, dalla tortuosità deicanali interni alle particelle e dal raggio delle stesse, Rp; ee rap-presenta la porosità esterna, C
_i la concentrazione media nella
particella, Cie la concentrazione nel cuore della particella.
La seconda equazione,
[131]
esprime invece la variazione della concentrazione interna espres-sa come differenza tra la velocità del trasferimento di materiaora considerata e la velocità di adsorbimento del componentesul solido; rs appresenta la densità del solido ed ei la porositàinterna.
Infine la terza equazione,
[132]
esprime il legame tra la concentrazione nei macropori e quel-la sul solido espressa dall’isoterma di adsorbimento; feqi
è infat-ti la grandezza che descrive l’equilibrio di adsorbimento.
Integrando numericamente le equazioni [130-132] è pos-sibile descrivere l’evoluzione nel tempo delle sostanze adsor-bite all’interno delle particelle.
Bilanci delle colonne di adsorbimentoLa letteratura è piuttosto ricca di studi sui criteri e le appros-
simazioni con cui si possono scrivere le equazioni che espri-mono il bilancio dinamico per le colonne di adsorbimento. Inun’impostazione generale si deve tenere presente che per lopiù si ha a che fare con sistemi a più componenti descritti dauna isoterma di adsorbimento non lineare. L’equazione
[133]
esprime il bilancio transitorio del componente i su un elementodi volume della colonna, tenendo conto degli effetti di disper-sione assiali e convettivi. La variabile u rappresenta la velocitàdel gas. Nel valutarlo si è tenuto conto della variazione di velo-cità del fluido dovuta alla variazione di portata che ha luogolungo la colonna a causa dei processi di adsorbimento. Que-sto effetto può diventare importante nella descrizione dei pro-cessi di separazione delle miscele concentrate nei quali, inseguito all’adsorbimento, può aver luogo un marcato impove-rimento di uno o più componenti della miscela. L’equazione[133] va quindi associata alle equazioni [131] e [132] di bilan-cio su una singola particella, e all’equazione di congruenzastechiometrica
[134]
Le equazioni semplificate sono le seguenti:
[135]
[136]
dove y è la frazione molare in fase gas, e è la frazione di vuo-to nella colonna, X
_la concentrazione media sul solido, X(y)
la concentrazione media sul solido in equilibrio con y, deff il
∂∂ = ( )− Xt R
X y Xeff152
δ
∂∂ + −( ) ∂
∂ =− ∂∂
yz
Xt
yts
s1 ε εr
M x Vi i
ii
n
r=∑ =
1
�
ε εeie
ie
Lie
ip
e iCt
uCz D C
zK R C∂
∂ +∂( )∂ − ∂
∂+ −( )
2
23 1 ee
iC−( )=0
ddt
f
CCt
i eq
j
j
j
ni
Γ=
∂
∂∂∂=
∑1
ε εei
ip
ie
i i sidC
dtK
RC C
ddt
= −( ) − −( )31 r
Γ
ε εei
ip
e ie
idCdt K K C C=− −( ) −( )3 1
b Ci i i jj
ni
jj
n0
1
1
1exp −
=
−=
=
∑∑
χ θ θ
θ
ASPETTI PROCESSISTICI
342 ENCICLOPEDIA DEGLI IDROCARBURI

coefficiente efficace di diffusione nel solido, z la coordinataassiale, R il raggio delle particelle.
La loro applicazione è agevolata dalla soluzione analiticaproposta da Rosen, per semplificazione delle soluzioni esatteproposte da Anzelius e Nusselt in un analogo problema di tra-sferimento di calore. Questa soluzione analitica viene ampia-mente usata, tuttora, in problemi di purificazione (Kast, 1981).
6.2.10 Scambio ionico
L’operazione detta di scambio ionico è costituita da uno scam-bio reversibile di ioni tra un solido e un liquido, che non pro-voca variazioni permanenti nella struttura del solido. Questotipo di operazione viene impiegata spesso in processi di addol-cimento e deionizzazione dell’acqua, ma costituisce altresì unmetodo di separazione estremamente utile in molti processichimici. La sua utilità risiede soprattutto nella possibilità diriutilizzare i materiali impiegati per eseguirlo.
Per esempio, nelle operazioni di addolcimento dell’acqua,avviene la reazione:
Il materiale R_
, che è solitamente una resina contenenteioni sodio, è in grado di scambiare lo ione calcio con la faseacquosa. In seguito la resina carica di calcio può essere trat-tata con una soluzione di cloruro di sodio, in modo da rige-nerarla nella forma originaria, per renderla disponibile per unnuovo ciclo di operazioni attraverso una reazione reversibiledi rigenerazione. In generale è possibile addolcire milioni dilitri d’acqua con 1 m3 di resina, operando per un periodo didiversi anni. Esistono diverse sostanze in grado di scambiareioni, per esempio silicati, fosfati, cellulosa. Lo scambio ioni-co, applicato industrialmente a partire dal 1910 circa, utilizzòdapprima zeoliti naturali; intorno al 1935 furono introdotteresine organiche sintetiche, costituite per esempio da polime-ri contenenti gruppi solfonici, carbossilici o fenolici, che pos-sono essere considerati come costituiti da un anione estrema-mente grande e da un catione scambiabile e che rendono age-voli scambi del tipo:
Na��HR��NaR �H�
In modo simile, resine polimeriche contenenti gruppi ammi-nici e anioni, possono essere utilizzate per scambiare anioni insoluzione:
RNH3OH �Cl���RNH3Cl �OH�
H��OH���H2O
dove RNH3 rappresenta la porzione cationica immobile dellaresina. Queste resine possono essere rigenerate per contattocon soluzioni di carbonato o idrossido di sodio. In generalesono disponibili resine sintetiche di scambio ionico di diversotipo, con varie capacità di scambio, solitamente sotto forma disolidi granulari.
Le tecniche operative abitualmente utilizzate per l’adsor-bimento sono impiegate anche per lo scambio ionico. L’ope-razione viene condotta generalmente utilizzando letti fissi diresina in cui si alternano la fase utile di scambio ionico e larigenerazione. Meno frequente è l’impiego di letti fluidizzati.Metodi cromatografici sono stati impiegati per il frazionamentodi miscele ioniche multicomponenti. Oltre ai metodi di addol-cimento dell’acqua citati sopra, la deionizzazione completadell’acqua può essere realizzata eseguendo una percolazione
prima attraverso una resina cationica e poi attraverso una resi-na anionica. Impiegando un letto formato da un’intima misce-la di quantità equivalenti di una resina cationica forte e unaresina anionica forte, è possibile rimuovere simultaneamentetutti gli ioni fino a raggiungere la neutralità. Le due resine devo-no poi essere separate � mediante classificazione idraulica,sfruttando le diverse dimensioni delle particelle e la loro den-sità � per poter essere rigenerate.
La distribuzione di equilibrio di uno ione tra una resina euna soluzione può essere descritta graficamente mediante curveisoterme del tutto simili a quelle impiegate per l’adsorbimentoordinario. Per descrivere queste isoterme si possono utilizzaresvariate equazioni empiriche, tra cui l’equazione di Freundlich.
D’altra parte le reazioni di scambio ionico sono reversibi-li. Lavando una resina con un eccesso di elettrolita, è possibi-le convertirla interamente alla forma ionica desiderata:
Tuttavia se la quantità di B� in soluzione è limitata, si sta-bilisce un equilibrio che dipende dalle proporzioni di A� e B�
e dalla selettività della resina. Il coefficiente di selettività perquesta reazione è dato da:
[137]
dove m e m_
si riferiscono rispettivamente alle concentrazioniioniche in soluzione e in fase solida.
La velocità alla quale avviene lo scambio ionico dipende,come per l’adsorbimento convenzionale, dalle velocità deglistadi in cui si articola il processo: a) diffusione degli ioni dalcuore della fase liquida alla superficie esterna di una particel-la della resina di scambio; b) diffusione degli ioni all’internodel solido verso il sito in corrispondenza del quale avviene loscambio; c) scambio degli ioni; d ) diffusione degli ioni rila-sciati verso la superficie esterna del solido; e) diffusione degliioni rilasciati dalla superficie del solido verso il cuore dellafase liquida.
In alcuni casi la cinetica dello stadio d) è determinante, maspesso essa è molto rapida rispetto alle velocità degli stadi dif-fusionali.
6.2.11 Sedimentazione e centrifugazione
Mediante sedimentazione è possibile separare, per effetto delcampo gravitazionale, una miscela eterogenea, costituita daparticelle solide sospese in un liquido, in due fasi distinte, ecioè in una fase solida, costituita da una sospensione in cui laconcentrazione delle particelle solide sospese è più elevata diquella della sospensione di partenza, e in una fase liquida, incui la concentrazione delle particelle solide sospese è moltopiù bassa. Lo scopo dell’operazione può essere quello di adden-sare la fase solida oppure di chiarificare la fase liquida. Unaparticella che si muove rispetto al liquido in cui è sospesa subi-sce una forza, detta di trascinamento
[138]
dove v è la velocità relativa particella-liquido, rL è la densitàdel liquido, A è la superficie della particella proiettata nelladirezione del moto e CD, detto coefficiente di trascinamento,è una funzione del numero di Reynolds
F C Av
D DL=
r 2
2
K mm
mmA
B B
A
A
B= ⋅
RA B RB A+ + + ++ +��➤�
2RNa Ca R Ca 2Na22
2+ + + ++ +��➤�
PROCESSI DI SEPARAZIONE
343VOLUME V / STRUMENTI

[139]
dove x rappresenta la dimensione della particella e m è la visco-sità del liquido. Il coefficiente CD dipende dal regime di flus-so. Generalmente le sedimentazioni avvengono in regime lami-nare, a basso numero di Reynolds (Rep�0,2). In queste con-dizioni CD può essere calcolato utilizzando la relazione:
[140]
Una particella di una sospensione, sottoposta al campo gra-vitazionale, è soggetta alla forza di trascinamento, alla forzapeso e alla spinta di Archimede, per cui vale la relazione:
[141]
dove m è la massa della particella, g l’accelerazione di gravità,rs la densità della particella e t il tempo. Quando si raggiun-gono le condizioni stazionarie (dv/dt�0), dalla [141] risulta
[142]
Nel caso di particelle sferiche di diametro d si ha:
[143]
Pertanto, inserendo nella [142] le relazioni [138], [140] e[143], si ottiene la legge di Stokes:
[144]
dove vg viene anche detta velocità terminale. In generale lavelocità terminale viene raggiunta rapidamente e la fase tran-sitoria può essere trascurata. L’equazione [144] è stata ricava-ta ipotizzando particelle isolate e può essere considerata vali-da per sospensioni sufficientemente diluite. Al crescere dellaconcentrazione le particelle si avvicinano tra loro e comincia-no a interferire reciprocamente, provocando un rallentamentonella sedimentazione che può essere espresso come segue:
[145]
dove vp è la velocità di sedimentazione della sospensione con-centrata, e è la frazione volumetrica del fluido e f(e) una suafunzione. Esistono diversi modi di esprimere f(e), tra cui il piùnoto è costituito dall’equazione di Barman-Kozeny:
[146]
Un modo piuttosto comune per far aumentare la velocitàdi sedimentazione vg consiste nell’aumentare la dimensionedelle particelle. Ciò viene generalmente ottenuto provocandoprocessi di aggregazione tra le particelle, detti anche di coa-gulazione, solitamente innescati aggiungendo additivi, peresempio polielettroliti. I sedimentatori industriali possono ope-rare in modo discontinuo o continuo. I primi sono molto sem-plici, ma relativamente poco utilizzati, a meno che si debbanotrattare delle piccole quantità di sospensioni.
Mediante la centrifugazione si separano i componenti di unamiscela, sottoponendola a un campo centrifugo la cui intensità
è data da w2r, dove w è la velocità angolare e r la distanza dal-l’asse di rotazione. Tale campo agisce in modo analogo al campogravitazionale (la cui intensità è g), ma la sua intensità puòessere variata cambiando la velocità di rotazione o le dimen-sioni dell’apparecchiatura, laddove il campo gravitazionale ècostante. Le apparecchiature industriali sono in grado di pro-durre accelerazioni che sono anche 20.000 volte quella di gra-vità, mentre le apparecchiature di laboratorio possono arriva-re fino a 300.000 g.
La centrifugazione viene soprattutto impiegata per sepa-rare dei componenti immiscibili o insolubili da un mezzo liqui-do. In generale la centrifugazione permette di eseguire conmaggiore efficacia le stesse operazioni che possono essere ese-guite sotto l’azione del campo gravitazionale. Diversi tipi diapparecchiature permettono di separare una miscela per cen-trifugazione, con varie configurazioni e geometrie. Sono cita-te le centrifughe a bottiglia, quelle tubolari e quelle a disco.
Nelle apparecchiature di centrifugazione si producono flus-si piuttosto complicati che hanno finora ostacolato lo svilup-po di un modello matematico che consenta un dimensiona-mento razionale. Un approccio semplificato consiste nell’uti-lizzare la legge di Stokes anche per il campo centrifugo. Siperviene così all’equazione:
[147]
dove vs è la velocità di caduta di una particella in un campocentrifugo, w è la velocità angolare della centrifuga, r è ladistanza dall’asse di rotazione a cui tale velocità si determina,vg è la velocità terminale della particella nel campo gravita-zionale, rs è la densità della particella, r la densità del mezzo,d il diametro della particella e m la viscosità del mezzo. Si sup-pone che la particella si trovi in una posizione iniziale corri-spondente a una distanza r dall’asse di rotazione. Applicandol’equazione [147] a questa particella, e assumendo vs�dr�dt,abbiamo:
[148]
dove rc è il raggio della torta sedimentata e t è l’intervallo ditempo durante il quale la particella subisce il campo centrifu-go. Integrando si ottiene:
[149]
L’equazione precedente è valida sotto una serie di ipotesisemplicative, in particolare:• se le particelle da cui è composta la sospensione sono sfe-
riche e tutte uguali e non cambiano forma o dimensioni,per effetto di coalescenze o flocculazioni, nel corso dellacentrifugazione;
• se le particelle sono uniformemente distribuite nella sospen-sione e la loro concentrazione è sufficientemente bassa dapotersi depositare come se fossero particelle isolate;
• se la velocità di caduta è tale che il numero di Reynolds èminore di 100, di modo che l’applicazione della legge diStokes non provochi un errore superiore al 10%.
6.2.12 Filtrazione
La filtrazione è l’operazione che consente di separare le parti-celle di una sospensione (liquida o gassosa), facendola passare
lnrr
vgtc
g= ω 2
drr
vgdtg
t
r
rc = ∫∫ω 2
0
vd r
v rgs
sg=
−( )=
r r 2 22
18
ω
µω
f ε εε( )= −( )10 1
vv fp
g= ( )ε ε2
vg d
gs=−( )r r
18
2
µ
m d
A d
s=
=
π
π6
4
3
2
r
gFms
D1−
=r
r
m dvdt
mg mg Fs
D≈ − −r
r
CDP
= 24
Re
RePLx
=νµr
ASPETTI PROCESSISTICI
344 ENCICLOPEDIA DEGLI IDROCARBURI

su un setto permeabile, detto anche filtro. Questo trattiene leparticelle solide e permette il passaggio del fluido. Procedendonella operazione, sul mezzo filtrante si accumula una torta diparticelle solide che costituiscono una massa porosa, com-primibile o meno. Nella torta le particelle trasmettono una sol-lecitazione meccanica mediante il loro contatto reciproco,detta anche pressione efficace. Se la torta, è incomprimibile,essa è dotata di una struttura rigida, e quindi non si deforma,neanche al crescere della pressione efficace. Se la torta è costi-tuita da particelle solide non flocculate, tipo sabbia o cristal-li di sale, essa può essere considerata non comprimibile, men-tre se le particelle che la compongono sono flocculate, comeavviene nei fanghi di scarto, negli idrossidi metallici o neiflocculi polielettrolitici, la torta è dotata di una struttura defor-mabile, e quindi comprimibile. La concentrazione dei solidiin una torta comprimibile è variabile e dipende dai valori loca-li di pressione efficace. Abitualmente in un processo di fil-trazione viene mantenuta una differenza di pressione idrauli-ca costante tra le camere ai due lati del filtro. La sospensionescorre attraverso il filtro, depositando le particelle solide sullasua superficie. La pressione applicata obbliga la sospensionea fluire anche attraverso la torta che via via si deposita sul fil-tro.
Di solito le particelle che formano il letto poroso sonomolto piccole, e quindi è piccolo anche il diametro medio deicanalicoli. Di conseguenza le velocità di passaggio del fluidoattraverso il filtro sono generalmente basse e il moto del flui-do è di tipo laminare. La caduta di pressione Dpa in un mezzoporoso si calcola mediante l’equazione di Blake-Kozeny:
[150]
dove e è la frazione di vuoto nel mezzo poroso, m è la visco-sità del fluido, Deq è il diametro equivalente delle particelle, Llo spessore del mezzo poroso e us è una velocità di passaggionel filtro definita come rapporto tra la portata volumetrica delfluido e l’area totale del filtro. Poiché in un letto solido otte-nuto da una sospensione di cristalli è praticamente impossibi-le determinare Deq ed e, è quindi opportuno scrivere la [150]nella forma seguente, detta equazione di Darcy:
[151]
Il coefficiente a, detto permeabilità, dipende dalle carat-teristiche del letto poroso e può essere determinato per viaempirica applicando direttamente l’equazione di Darcy. Rica-vando da essa us, si ottiene:
[152]
da cui
[153]
essendo A la sezione totale del filtro e dV il volume di liquidoche filtra nel tempo dt.
Dopo un tempo t, se V è il volume del liquido filtrato, L lospessore della torta solida, rs la densità del solido che la costi-tuisce e r quella del liquido, da un semplice bilancio materia-le si ottiene la relazione:
[154]
dove e è al solito la porosità del setto poroso, x la frazione disolido nella sospensione. Risolvendo la [154] rispetto a V e aL si ottiene:
[155]
[156]
Sostituendo la [155] nella [153], si ha:
[157]
dove Kv indica una costante che ingloba tutte le proprietà carat-teristiche della massa porosa e dell’alimentazione, espressa da:
[158]
Se la porosità e la permeabilità si mantengono costantidurante la filtrazione, circostanza che si verifica per torte incom-primibili, Kv è pure costante. Inoltre se anche Dpa è costantela [158] si integra facilmente:
[159]
definendo
[160]
È possibile ricavare una relazione che leghi lo spessoredella torta solida al tempo t scrivendo la [156] in forma diffe-renziale e sostituendola nella [153]:
[161]
Introducendo la costante KL definita dalla relazione
[162]
la [161] diventa:
[163]
Se la differenza di pressione è costante, la [163] può esse-re facilmente integrata fornendo:
[164]
Le costanti Kv e KL possono essere calcolate dalle pro-prietà dell’alimentazione e della torta solida. In pratica peròtale valutazione è particolarmente difficile, soprattutto perquanto riguarda la stima della porosità della torta e della formae delle dimensioni delle particelle che la costituiscono; si pre-ferisce quindi ricorrere a una valutazione sperimentale diret-ta che utilizza le equazioni [157] e [164], misurando il volu-me di liquido filtrato e lo spessore della torta solida in fun-zione del tempo.
Nel ricavare le equazioni precedenti è stato ipotizzato chela resistenza incontrata dal fluido sia dovuta unicamente allatorta solida. In realtà bisogna tener conto che esistono altrefonti di resistenza, in primis il setto poroso e poi l’insieme dicondotti, connessioni e valvole che costituiscono il filtro. Que-sto insieme di resistenze può essere espresso mediante uno
t K LpL
a=
2
∆
dLdt
pK L
a
L= ∆
2
Kx x
xLs=−( ) −( )− µ ε εα
r rr
1 1
2
dLdt
x pL x x
a
s
=−( ) −( )−
αµ ε ε
r
r r
∆
1 1
t K VA p
v
a
=2
2∆
22
00
KA p
VdV dtv
a
tV
∆= ∫∫
K xx xv
s
=−( ) −( )−
µα ε ε
r
r r2 1 1
dVdt
A x x pV x
A ps a a=−( ) −( )− =
α ε εµ
2 21 12
r rr
∆ ∆KK Vv
L V xA x xs
=−( ) −( )−
r
r r 1 1 ε ε
Vx x
x LAs=−( ) −( )−r r
r 1 1 ε ε
1 1−( ) =+( )−ε ε
LAV LA x
xrr
dVdt
p ALa=αµ
∆
u AdVdt
pLs
a= =1 αµ∆
∆pL ua
s= 1α µ
∆pL
u
Da s
eq
=−( )
( )1 1502
3 2εε
µ
PROCESSI DI SEPARAZIONE
345VOLUME V / STRUMENTI

‘spessore equivalente della torta’ Leq e un ‘volume equivalen-te di filtrazione’ Veq.
Le equazioni [160] e [163] assumono quindi la forma:
[165]
[166]
Integrando a Dpa costante si ottiene:
[167]
[168]
La [165] si può scrivere nella forma:
[169]
Determinando sperimentalmente la serie di valori di DV,frazioni di liquido filtrato in diversi intervalli di tempo Dt, sesi riporta in un grafico in funzione di V, si ottiene una retta rap-presentativa dell’equazione [169]. La pendenza della retta for-nisce il valore di 2Kv �A2Dpa, mentre l’intercetta sull’asse delleascisse dà il valore di 2KvVeq �A2Dpa. Noti A e Dpa si possonoquindi determinare Kv e Veq.
Se la torta è comprimibile, la sua porosità e dipende dallapressione e perciò Kv varia con Dpa. Inoltre particelle di soli-do possono entrare e bloccare gli interstizi del setto, variandoVeq in un modo che dipende anch’esso da Dpa.
6.2.13 Processi di separazione a membrana
È possibile ottenere la separazione di miscele fluide omoge-nee utilizzando membrane che permettono il passaggio di unoo più componenti, mentre ritardano il passaggio di altri. Lacomposizione ai due lati della membrana è quindi differente.Esiste una grande varietà di processi a membrana e in un certosenso anche la filtrazione, che consiste nella separazione diparticelle grosse e visibili da un liquido o da un gas, può esse-re considerata un tipo particolare di separazione a membrana.Al capo opposto dello spettro rientrano quei processi che per-mettono di separare ioni o molecole sulla base del loro pesomolecolare. I processi a membrana possono essere classifica-ti sulla base delle dimensioni delle particelle o delle molecole
che vengono separate (tab. 4). I vari meccanismi che possonointervenire nelle separazioni sono elencati di seguito.
Meccanismo di esclusione dimensionale. Le membranesono dotate di pori che permettono il passaggio di alcune spe-cie e non di altre.
Meccanismi di diffusione di Knudsen. Le membrane sonoprovviste di pori le cui dimensioni sono vicine a quelle mole-colari, causando dei ritardi selettivi nel passaggio di alcune spe-cie. Questo processo avviene soprattutto nelle separazioni insistemi gassosi, in cui membrane con pori molto piccoli pos-sono indurre separazioni legate a questo tipo di meccanismi,nei quali le velocità di diffusione delle varie molecole varianocome l’inverso della radice quadrata del loro peso molecolare.
Meccanismo di diffusione per soluzione. È possibile chealcune specie si sciolgano nella membrana, migrando succes-sivamente attraverso di essa per diffusione molecolare, rie-mergendo poi dall’altro lato.
L’importanza industriale e la rilevanza economica dei pro-cessi a membrana è crescente, con applicazioni che vanno daltrattamento delle acque alle industrie alimentare e farmaceu-tica.
I processi a membrana hanno una lunga serie di pro e con-tro, se confrontati con i processi più convenzionali. Tra i lorovantaggi più significativi bisogna citare i seguenti: • non richiedono variazioni di fase, e quindi comportano una
bassa spesa energetica;• gli schemi di processo sono molto semplici, con poche
apparecchiature ausiliarie;• talvolta sono efficaci in casi in cui i sistemi convenziona-
li falliscono, come nella separazione di miscele gassoseche formano azeotropi;
• non provocano significativi danni all’ambiente;• esiste una ampia scelta di membrane e quindi è possibile
esercitare un ottimo controllo sulle selettività del processo.Esistono però anche delle condizioni in cui questi proces-
si sono difficilmente applicabili, per esempio nel caso in cuivi sia incompatibilità chimica tra la membrana e il sistema datrattare, oppure nel caso in cui possano formarsi degli sporca-menti sulla superficie della membrana che diminuiscono sen-sibilmente la potenzialità del processo. Inoltre non si possonousare temperature elevate che danneggino la membrana.
Solitamente si impiegano due diverse configurazioni diflusso, perpendicolare o parallelo alla membrana. Nel primocaso si provoca un accumulo di materiale sulla superficie dellamembrana e l’operazione deve essere periodicamente inter-rotta per consentire il lavaggio. Nel secondo caso la possibi-lità di formazioni di accumuli diminuisce, perché il materialedepositato tende a essere trascinato via dal flusso.
dtdV
KA p
V KA p
Vv
a
v
aeq= +2 2
2 2∆ ∆
tC L LL
pL eq
a=
+( )2 2∆
tK V VV
A pv eq
a
=+( )2
2
2∆
dLdt
pK L L
a
L eq
=+( )
∆2
dVdt
A pK V V
a
v eq
=+( )
2
2∆
ASPETTI PROCESSISTICI
346 ENCICLOPEDIA DEGLI IDROCARBURI
tab. 4. Classificazione dei processi a membrana
Processo Meccanismo di separazione Dimensione dei pori (Å) Regime di trasporto
Filtrazione Esclusione dimensionale �50.000 Macropori
Microfiltrazione Esclusione dimensionale 500-50.000 Macropori
Ultrafiltrazione Esclusione dimensionale 20-50 Mesopori
Nanofiltrazione, osmosi inversaEsclusione dimensionale;
soluzione/diffusione�20 Micropori
Separazione di gas Soluzione/diffusione �5 Molecolare

Le forze motrici che agiscono nelle separazioni con mem-brana possono essere di vario tipo: differenza di pressione, gra-diente di concentrazione, differenza di potenziale elettrico.
Il flusso NA di un componente A attraverso una membra-na è dato dall’equazione:
[170]
dove PA è la permeabilità di A, L è lo spessore della membra-na e D/A la forza motrice che induce il passaggio attraverso lamembrana. Per le separazioni in fase gassosa, D/A è data daDpA, che è la differenza tra le pressioni parziali di A ai due latidella membrana. Per le separazioni in fase liquida la situazio-ne è più complessa. Per un liquido puro D/A sarebbe banal-mente dato dalla differenza di pressione ai due lati della mem-brana, p1�p2 ma, aggiungendo una piccola quantità di solutoincapace di attraversare la membrana, può insorgere una ridu-zione del flusso indotta da effetti dovuti alla pressione osmo-tica p. Il flusso di A infatti in questo caso è dato da:
[171]
Gli effetti osmotici possono essere trascurati nelle separa-zioni di specie con pesi molecolari superiori a qualche migliaiodi dalton, ma se il soluto ha un basso peso molecolare essi pos-sono diventare sorprendentemente rilevanti e tali da limitarela capacità di separazione. Per concentrazioni di soluto suffi-cientemente basse la pressione osmotica può essere espressamediante l’equazione di van’t Hoff:
[172]
dove CA è la concentrazione del soluto, R è la costante univer-sale dei gas e T la temperatura termodinamica. L’effetto dellapressione osmotica è quello di opporsi all’aumento della con-centrazione del soluto, ovvero quello di impedire l’ottenimen-to di una corrente di liquido puro. Si noti che, per i materialia basso peso molecolare, la pressione osmotica è molto gran-de e si oppone alla concentrazione delle specie che non attra-versano la membrana. Per esempio, per pesi molecolari nel-l’ordine di 100 dalton, se la differenza di pressione è pari a 35bar, la massima concentrazione del soluto che è possibile otte-nere è pari circa al 22%. A questa concentrazione la pressioneosmotica è uguale alla differenza di pressione ai due lati dellamembrana e il flusso si annulla.
Per valutare la selettività di una membrana è possibile ricor-rere all’equazione [170]. Se la si scrive per una specie A e unaspecie B e si dividono le due equazioni così ottenute membroa membro, si ottiene:
[173]
Se DpA�DpB, la selettività della membrana è dato dal rap-porto delle permeabilità.
Applicando una differenza di potenziale elettrico ai due latidi una membrana, è possibile provocare il trasporto degli ioni.Questo tipo di processo viene denominato elettrodialisi e con-sente di separare o concentrare sali, oppure acidi e basi da solu-zioni. Le membrane utilizzate nell’elettrodialisi possono esse-re di tipo anionico o cationico. Entrambe sono generalmentecostituite da polistirene reticolato, ma nelle prime questo è statosuccessivamente solfonato, mentre nelle seconde esso contie-ne dei gruppi ammonici quaternari. Le prime permettono soloil passaggio degli anioni, le seconde solo quello dei cationi.
6.2.14 Flottazione
Nella separazione per flottazione viene preparata una sospen-sione del materiale da trattare, a cui vengono aggiunti com-posti chimici, detti agenti collettori, che hanno il potere direndere idrofoba la superficie del materiale. Insufflando aria,le bolle di gas rimangono attaccate alle particelle di minera-le che, pertanto, risalgono in superficie formando una schiu-ma. La flottazione è ampiamente applicata nell’arricchimen-to dei minerali. La superficie dei minerali ha solitamente carat-teristiche polari e quindi idrofile; si rende pertanto necessarial’aggiunta di agenti collettori che riducono notevolmente l’af-finità delle particelle di minerali con l’acqua. I collettori piùcomuni sono sali organici con lunghe catene idrocarburiche.La parte polare si orienta verso la superficie del minerale, chequindi esternamente risulta protetto da uno strato idrofobo eviene trascinato dalle bolle d’aria. Il processo in questione ècontrollato da molti fattori, tra cui il pH, le dimensioni e iltipo della superficie da trattare. I collettori possono essere ditipo anionico o cationico, a seconda se la carica che essi por-tano è positiva o negativa. Gli anionici si distinguono in ossi-drilici e solfidrilici. I collettori cationici sono in genere saliammonici quaternari e si usano per i minerali con forte den-sità di carica superficiale. Nelle operazioni di flottazione siusano anche agenti deprimenti, sostanze che modificano lasuperficie del materiale rendendola più idrofila, impiegatisoprattutto nella flottazione selettiva di due minerali che hannoaffinità per lo stesso agente collettore. Tali agenti si usano perdiversificare la superficie dei minerali che possono essereseparati in due stadi di flottazione. Un’altra classe di additi-vi utilizzati nelle flottazioni è quella degli schiumeggiatori,che servono a coadiuvare la formazione di schiume stabili ea favorire l’adesione delle bolle d’aria al complesso minera-le-collettore.
In pratica, la flottazione viene eseguita sul materiale maci-nato e disperso in acqua; quest’ultima viene immessa in unacella verticale dentro cui viene generata una corrente di minu-tissime bolle d’aria. Il numero di celle di flottazione varia infunzione del numero e delle specie di minerali presenti.
Bibliografia generale
King C.J. (1983) Separation processes, New York, McGraw-Hill.
McCabe W.L. et al. (2005) Unit operations of chemical engineering,Boston (MA), McGraw-Hill.
Seader J.D., Henley E.J. (2005) Separation process principles,Etobicoke (Canada), John Wiley.
Bibliografia citata
Broughton D.B. et al. (1970) Parex process for recovering paraxylene,«Chemical Engineering Progress», 66, 70-75.
Fenske M.R. (1932) Fractionation of straight-run Pennsylvaniagasoline, «Industrial and Engineering Chemistry», 24, 482-485.
Gilliland E.R. (1940) Multicomponent rectification. Minimum refluxratio, «Industrial and Engineering Chemistry», 32, 1101-1106.
Kast W. (1981) Adsorption aus der gasphase - Grundlagen unVerfahren, «Chemie Ingenieur Technik», 53, 160-172.
McCabe W.L., Thiele E.W. (1925) Graphical design of fractionatingcolumns, «Industrial and Engineering Chemistry», 17, 605-611.
Miers H.A. (1906), «Journal of the Chemical Society», 89, 413.
NN
PLPL
pp
PP
pp
A
B
A B A
B
A
B
A
B
= =∆∆
∆∆
πA AC RT=
NPL
p pAA
A= − −( )1 2π
NPLAA
A= ∆φ
PROCESSI DI SEPARAZIONE
347VOLUME V / STRUMENTI

Murphree E.V. (1925) Rectifying column calculations with particularreference to n-component mixtures, «Industrial and EngineeringChemistry», 17, 747-750.
Ostwald W. (1896) Lehrbuch der Allgemeinen Chemie, Leipzig,Engelmann, 2v.; v.II, Part 1.
Randolph A.D., Larson M.A. (1962) Transient and steady-state sizedistributions in continuous mixed suspension crystallizers,«American Institute of Chemical Engineers Journal», 8, 639-645.
Seko M. et al. (1979) Economical p-xylene and ethylbenzene separatedfrom mixed xylene, «Engineering Chemistry Process Design andDevelopment», 18, 263-268.
Underwood A.J.V. (1948) Fractional distillation of multicomponentmixtures, «Chemical Engineering Progress», 44, 603-614.
Elenco dei simboli
a superficie di contatto liquido/gas per volume di gasin una colonna di assorbimento
a superficie per volume unitario del solido, inun’operazione di essiccamento
A sezione trasversale di una colonna di assorbimentoA massa del diluente nella corrente di alimentazione
di un estrattore liquido-liquidoA superficie della particella proiettata nella direzione
del motoA superficie di un cristalloB massa del solvente puro presente nella corrente del
solvente in un’estrazione liquido-liquidobi parametri dell’equazione di LangmuirCD coefficiente di trascinamentoC1 concentrazione della specie 1 nel cuore della fase
liquidaC1i concentrazione della specie 1 in corrispondenza
della superficie interfasicaC1
* concentrazione che si avrebbe nel liquido se fossein equilibrio con p1
Ci concentrazione di i nella fase fluidaCi� concentrazione del soluto all’interfaccia con il
solido in una cristallizzazioneCi
e concentrazione media nel cuore della fase fluida diuna colonna di adsorbimento
C_
i concentrazione media nelle particelle di unacolonna di adsorbimento
Cs concentrazione del soluto nel cuore della soluzione,in un cristallizzatore
cs calore specifico dell’aria umidad diametro di una particellaD portata molare del prodotto di testa di una colonna
di distillazioneD coefficiente di diffusione del componente che
cristallizza in soluzioneDeq diametro equivalente delle particelleDi coefficiente di diffusione del componente iDl coefficiente di diffusione in un liquidoE portata molare per unità di sezione unitaria della
fase estratta in un estrattore continuoEME efficienza di un’operazione di estrazione liquido-
liquido, riferita all’estrattoEMR efficienza di un’operazione di estrazione liquido-
liquido, riferita al raffinatoEMV efficienza di MurphreeEs portata di un solvente puro
F portata molare dell’alimentazioneFD forza di trascinamentoF(e) portata massiva all’ingresso di un’apparecchiaturaF(u) portata massiva all’uscita di un’apparecchiaturaFi
(e) portata massiva del componente i all’ingresso diun’apparecchiatura
Fi(u) portata massiva del componente i all’uscita di
un’apparecchiaturaFn alimentazione sullo stadio nFe
(i) portata massiva del componente i all’ingresso diun’apparecchiatura
Fu(i) portata massiva del componente i all’uscita di
un’apparecchiaturaF(u) portata molare in uscita da un’apparecchiaturag accelerazione di gravitàG velocità di crescita dei cristalli Gs portata molare del gas insolubile per sezione unitaria
di colonna Hi costante di Henry H umidità assoluta HA umidità percentualeHR umidità relativaHs umidità assoluta in condizioni di saturazione HOG altezza di una unità di trasferimento, nelle operazioni
di assorbimento di gas HOR altezza di una unità di trasferimento, nelle operazioni
di estrazione liquido-liquido H entalpia molare del vaporeh coefficiente di trasferimento di calore da gas a liquido h entalpia molare del liquido hF entalpia molare dell’alimentazione Ki rapporto di vaporizzazione del componente iK energia cinetica riferita all’unità di massa kc coefficiente di trasporto di materia, in cristallizzazione kg coefficiente di trasporto di materia in fase gasKg coefficiente globale di trasporto di materia in fase
gaskl coefficiente di trasporto di materia in fase liquidaKl coefficiente globale di trasporto di materia in fase
liquidaL portata di liquidoL lunghezza caratteristica dei cristalliL spessore della membranaLeq spessore equivalente della tortaLD portata molare del componente condensato rinviato
in colonnaLs portata molare del solvente puro per la sezione
unitaria di colonnaLn portata molare del liquido che scende dal piatto ml spessore in un solido sottoposto a essiccamentom massa di solido che viene depositata nel corso di
una cristallizzazionemi massa del componente i contenuta in
un’apparecchiaturaN numero totale di cristalli contenuti in un
determinato volumeN moli del componente solubile trasferito per unità di
tempo dal gas al liquidoNA flusso di un componente A attraverso una
membranaNm numero minimo di stadi di una colonna
ASPETTI PROCESSISTICI
348 ENCICLOPEDIA DEGLI IDROCARBURI

NOR numero di unità di trasferimentoN1 moli della specie 1 trasferite per unità di tempo e
per unità di superficie di contatton numero di cristalli per volume unitario aventi una
determinata lunghezzani numero di moli del componente i contenute nel
sistemaP pressione totalePA permeabilità di Ap pressione parziale dell’acquaps tensione di vapore dell’acquapi
0 tensione di vapore del liquidop1i pressione parziale del componente 1 all’interfasep1
* pressione parziale che si avrebbe nel gas se fosse inequilibrio con C1
Q calore scambiato da un sistema termodinamicoQn calore scambiato sullo stadio nr velocità globale del processo di cristallizzazioner distanza dall’asse di rotazione a cui si determina la
velocità in un campo centrifugor raggio di un nucleo di cristallizzazioneR rapporto di riflussoR portata molare totale per sezione unitaria della fase
raffinatarc raggio critico di un nucleo di cristallizzazioneRm minimo rapporto di riflussoRp raggio delle particelle in una colonna di
adsorbimentoRep numero di Reynolds di una particella in una
sedimentazioneSij selettività di una faseTs temperatura della superficie liquidaU energia interna di un sistema riferita all’unità di
massa Un liquido prelevato dallo stadio nus velocità di passaggio nel filtroV portata di vaporeV volume per massa unitariaVeq volume equivalente di filtrazioneVm portata molare del vapore che sale dal piatto mv portata volumetrica del flusso uscente da un
cristallizzatorev velocità relativa particella-fluidovg velocità terminalevp velocità di sedimentazione della sospensione
concentratavs velocità di caduta di una particella in un campo
centrifugoW lavoro termodinamicoW lavoro isotermo di formazione di un nucleo di
cristallizzazioneW portata molare del prodotto di codaW umidità di un solidoWc umidità di un solido al punto criticoWe umidità di equilibrio in un solidown vapore prelevato dallo stadio nX rapporto tra massa di sostanza e massa di solvente
in un’estrazione liquido-liquidox dimensione di una particella in una sedimentazione
xi frazione molare del componente i in fase liquidaxn composizione del liquido che lascia il piatto nxE frazione molare nella fase raffinata del componente
che viene estrattoxR frazione molare nella fase raffinata del componente
che viene estrattoyi frazione molare del componente i nella fase vaporeY concentrazione nel gas del componente solubileY rapporto tra massa di sostanza e massa di solvente
in un’estrazione liquido-liquidoyn composizione del vapore che lascia il piattoyn
* composizione in equilibrio con la composizione xndel liquido che lascia il piatto
z quota della sezione di un condottozi frazione molare di i nell’alimentazioneZ altezza totale di una colonna di assorbimento
necessaria per realizzare una certa separazione
Lettere grechea coefficiente di permeabilità di un filtroas
ij fattore di separazione dei componenti i e jci parametro di interazione tra le specie adsorbitegi coefficiente di attività del componente iGi concentrazione di i nella fase solida di una colonna
di adsorbimentoGi
capacità di carico dell’adsorbented spessore dello strato liquido dove di è localizzata la
resistenza alla diffusione nella fase diaccrescimento di una cristallizzazione
D differenza tra i valori di una corrente entrante e unauscente
Dp caduta di pressione in una colonna di adsorbimentoe frazione volumetrica del fluidoe frazione di vuoto nel mezzo porosoee porosità esterna nelle colonne di adsorbimentoei porosità interna nelle particelle delle colonne di
adsorbimentoÿ tempo di residenza medio nel cristallizzatoreÿ parametro dell’equazione di Underwoodqi grado di ricoprimento della superficie da parte del
componente i.l calore di evaporazione dell’acquam viscosità della fase liquidarl densità del liquidors densità del solidorv densità del vapores tensione superficialew velocità angolare della centrifugaF energia potenziale
Sergio Carrà
Dipartimento di Chimica, Materiali eIngegneria chimica ‘Giulio Natta’
Politecnico di MilanoMilano, Italia
Stefano Carrà
MAPEI
Milano, Italia
PROCESSI DI SEPARAZIONE
349VOLUME V / STRUMENTI



















